
Abstract
Background:
Kaeng Khoi virus (KKV), which belongs to the genus Orthobunyavirus, family Perbunyaviridae, was originally isolated from the brain tissue of bats and may cause infection in humans. In this study, the KKV strain WDBC1403 was isolated from bat flies (Eucampsipoda sundaica), ectoparasites of the bat Rousettus leschenaultia, collected from Yunnan Province of China at the Sino-Burmese border.
Methods and Results:
The bat fly specimens were ground and inoculated in culture cells. The WDBC1403 strain was shown to induce cytopathic effects in Vero, baby hamster kidney (BHK-21), and Tb1Lu cells, but not in C6/36 cells; however, viral gene amplification was detected in the supernatants of C6/36 cells. Using electron microscopy, the virus was determined to be spherical, enveloped, and 80–90 nm in diameter; it was also shown to form plaques in BHK-21 cells and the titer reached 1 × 106.57 plaque-forming units/mL 24 h after infection. The phylogenetic analysis showed that WDBC1403 is a KKV strain, but is independent of the original KKV strain (PSC-19). Viral genome analysis revealed that the nucleotide and amino acid sequence identities of the S, M, and L segments of WDBC1403 with PSC-19 were 88.2% and 96.1%, 76.7% and 85.0%, and 78.3% and 88.9%, respectively. Two amino acids were removed at the end of the open reading frame of the M segment, and 47 nucleotides were removed in the 3′-untranslated region of the M segment of the WDBC1403 strain compared with the PSC-19 strain.
Conclusions:
The WDBC1403 strain is a highly divergent KKV strain, suggesting that there are a variety of KKV strains that exhibit molecular differences. Moreover, because of the large variations in nucleotide and amino acid sequence in the M segment, which encodes an important membrane protein, further research on antigenicity and pathogenicity in humans and animals is needed.
Introduction
T
WDBC1210 isolated from hematophagus bat fly (Eucampsipoda sundaica) specimens on the surface of bats collected in the Yunnan Province of China in 2012 was identified as a strain of KKV (Feng et al. 2017). Viral genome sequencing analysis revealed that the nucleotide and amino acid identities of the S, M, and L segments of WDBC1210 with PSC-19 are 96.9% and 98.7%, 94.1% and 97.0%, and 95.2% and 98.4%, respectively. These results suggested that although WDBC1210 was isolated ∼40 years after PSC-19 (1969–2012) and at a site ∼1400 km away, the genomes of the two KKV strains have maintained relatively similar molecular characteristics. Thus, it appears that KKV is not affected by natural geographical factors such as time and region, and that KKV has relatively stable genomic characteristics (Feng et al. 2017).
In the virus monitoring survey at the Sino-Burmese border in 2014, the strain WDBC1403 was isolated from bat fly (E. sundaica) specimens on the surface of bats collected in the same location from which WDBC1210 was previously isolated. Analysis revealed differences in the biological phenotype and molecular genetic features of this strain with the KKV PSC-19 strain and the WDBC1210 strain previously isolated from the same location.
Materials and Methods
Ethics statement
This research, including the procedures and protocols of specimen collection and processing, was reviewed and approved by the Medical Ethics Committee of the Yunnan Institute of Endemic Diseases Control and Prevention.
Sample collection
The nets for bat collection were arranged at an orchard in Wanding, Ruili, Dehong prefecture of Yunnan Province. The captured bats were carefully removed from the nets, and active bat fly specimens from the body hair of the bats were collected with tweezers, stored in cryopreserved tubes after morphological classification, and transported to the laboratory in liquid nitrogen for further analysis (Feng et al. 2017). The bats were released after the bat flies were collected. The bat fly specimens were identified as E. sundaica (Feng et al. 2017) by mitochondrial cytochrome oxidase I gene detection (Folmer et al. 1994, Feng et al. 2017).
Cell culture
Aedes albopictus (C6/36), African green monkey kidney (Vero), baby hamster kidney (BHK-21), and Tadarida brasiliensis lung (Tb1Lu) cells lines used in this study were stored by our laboratory. The C6/36 cell line was cultured with minimum essential medium (MEM; Gibco, Grand Island, NY) containing 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA) and 100 U/mL penicillin–streptomycin at 28°C (Wang et al. 2011). Vero, BHK-21, and Tb1Lu cell lines were cultured with MEM containing 10% FBS and 100 U/mL penicillin–streptomycin in a humidified incubator at 37°C with 5% carbon dioxide (CO2) (Wang et al. 2011, 2015, Feng et al. 2017).
Virus isolation
Ten bat fly specimens were placed in a tissue grinder, rinsed with 2 mL of MEM, and 1 mL of MEM containing 100 U/mL penicillin–streptomycin was added. The specimens were ground manually to homogenize and were centrifuged at 16,000 × g at 4°C for 20 min. Then, BHK-21 and Vero monolayer cells were inoculated with 0.2 mL of the supernatant, which was adsorbed at 37°C for 1 h. Then, 1 mL of the culture medium was added and cells were incubated at 37°C. Cells were observed daily. A virus isolate was considered positive when cytopathic effect (CPE) was induced in three sequential passages. When 75% of the cells exhibited marked CPE, the CPE level was considered as +++. The supernatants of positive isolates were collected after three freeze–thaw cycles and stored at −80°C for further analysis (Wang et al. 2011, Lei et al. 2015, Feng et al. 2017).
Electron microscopy
BHK-21 cells infected for 16 h were harvested, centrifuged, and fixed with 2% formaldehyde and 2.5% glutaraldehyde for conventional sample preparation into ultrathin sections. Epoxy resin PON812 was used to embed the polymerized samples for ultrathin (80 nm) sectioning. Sections were observed with electron microscopy after conventional dyeing (Lei et al. 2015).
Plaque assay
Virus suspensions were diluted with a 1:10 gradient ranging from 10−1 to 10−6, added to six-well plates of single-layer BHK-21 cells at 80% confluence, with two wells for each dilution (0.1 mL/well), and absorbed in a humidified incubator at 37°C and 5% CO2 for 1 h. Cells were then supplemented with 1.3% methyl cellulose–MEM containing 2% FBS (5 mL/well) and cultured continuously for 2 to 3 days. Crystal violet staining was performed once obvious plaques were observed under a microscope and the number of plaque-forming units (pfu) was calculated. The diameter of eight plaques in each well was measured and the mean and standard deviation were calculated (Cao et al. 2016, Feng et al. 2017).
Measurement of virus multiplication
BHK-21 cells at 80% confluence were inoculated with a virus suspension at a multiplicity of infection (MOI) of 0.1 and cultured in a humidified incubator at 37°C and 5% CO2. One tube was collected every 8 h after infection until 72 h and stored at −40°C. Subsequently, the virus titer was determined using a plaque forming test and a virus proliferation curve was generated (Cao et al. 2016).
Complete genome sequencing including 5′- and 3′-untranslated regions
RNA extraction with the QIAamp Viral RNA Mini-Kit (Qiagen, Hilden, Germany) was performed with 200 μL of the virus supernatant. The Ready-to-Go You Prime First-Strand Beads Kit (GE Healthcare, Little Chalfont, UK) was used to prepare a viral genome complementary DNA (cDNA) library. WDBC1210 primers were initially used for PCR amplification of the viral genome of WDBC1403 (Feng et al. 2017). Primers were then designed based on the obtained sequences, and the Genome Walking Kit (Takara, Japan) and the 5′/3′ rapid amplification of cDNA ends (RACE) system (Invitrogen) were used to amplify the whole genome sequence, including the 5′- and 3′-untranslated regions (UTRs). For the 3′-UTR without a polyA tail, the Poly(A) Polymerase Tailing Kit (Epicentre Biotechnologies, Madison, WI) was used for the addition of a poly(A) tail and amplified. The amplified products were detected with 1% agarose gel electrophoresis, purified with the QIAquick Gel Extraction Kit (Qiagen), and cloned into the pGEM-T Easy Vector (Promega, Madison, WI) for Sanger sequencing (Wang et al. 2011, Lei et al. 2015, Feng et al. 2017).
Primers were then designed based on the whole-genome sequence of WDBC1403 obtained with the mentioned method and a second amplification was performed. Sequencing of the S, M, and L genome segments with the viral cDNA library as a template was performed to confirm the genomic sequences (Supplementary Table S1; Supplementary Data are available online at
Sequence and phylogenetic analysis
The DNASTAR software package (Lasergene) was used for the analysis and assembly of viral nucleotide and deduced amino acid sequences. The NCBI database (
MEGA (version 7.0.18) software was used to construct the maximum-likelihood evolutionary tree of the whole-genome nucleotide sequences of the S, M, and L segments, or partial nucleotide sequences of the M segment, using the p-distance and the Poisson correction algorithms. The robustness of branching was evaluated by bootstrapping using 1000 replications (Wang et al. 2011, Lei et al. 2015, Cao et al. 2016, Feng et al. 2017).
Results
Isolation and biological characterization of the WDBC1403 strain
Vero and BHK-21 cells were inoculated with the supernatant of ground bat fly specimens for continuous culture. The WDBC1403 inoculum induced CPE characterized by the round shrinkage and exfoliation of both cells (Fig. 1). CPE was still observed in the third passage of both cells. Electron microscopy of ultrathin sections revealed spherical enveloped particles with a diameter of 80–90 nm in the cytoplasm of BHK-21 cells (Fig. 2A).
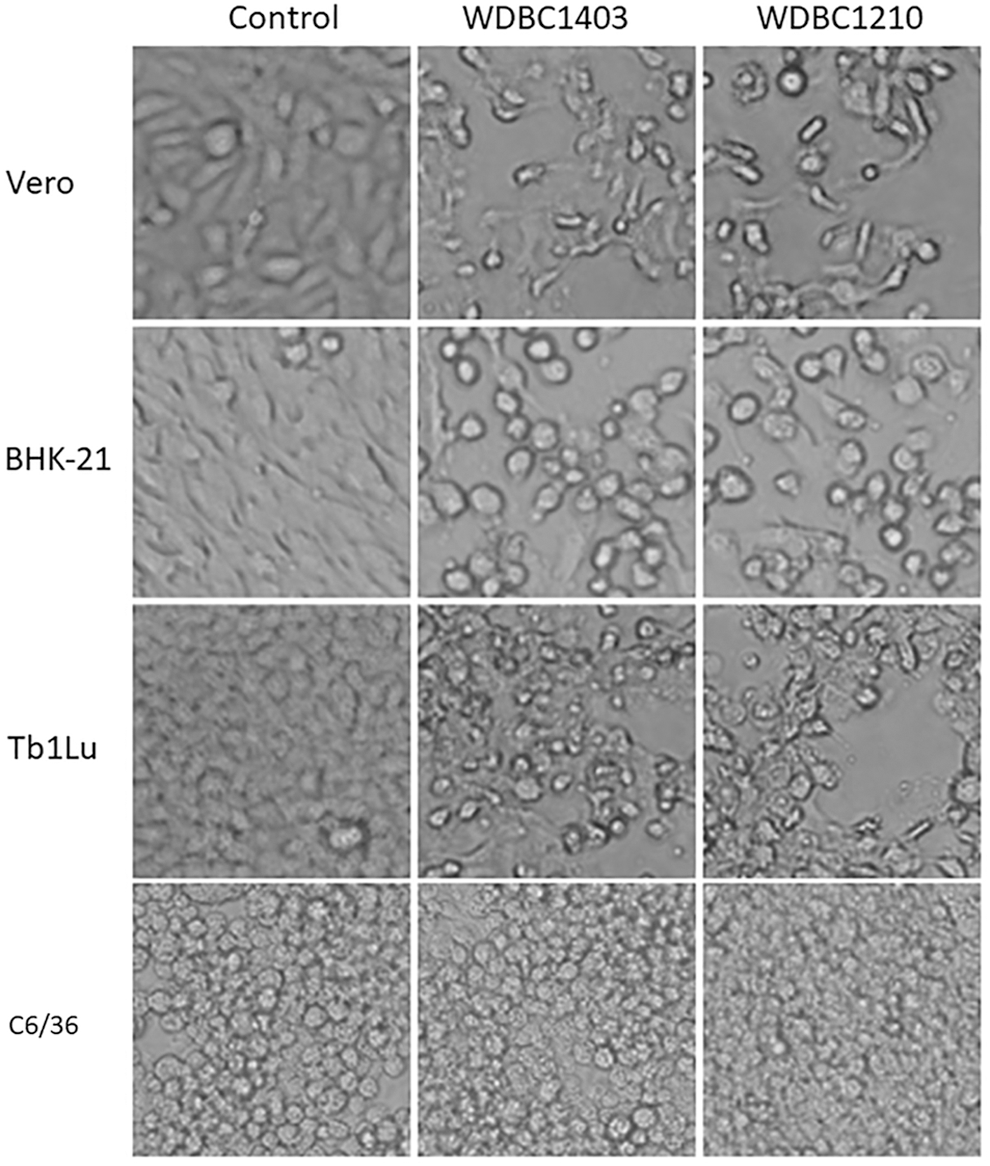
CPE induced by the strains WDBC1403 and WDBC1210. CPE induced in Vero, BHK-21, and Tb1Lu cells at 40 h, and C6/36 cells at 88 h, after inoculation of both strains (200 × ). BHK-21, baby hamster kidney; CPE, cytopathic effect.

Electron microscopy and analysis of the biological characteristics of the WDBC1403 strain.
A parallel observation was carried out with the inoculation of the WDBC1403 and WDBC1210 strains in cultured cells (C6/36, Vero, BHK-21, and Tb1Lu). CPE appeared at 24 h after inoculation in Vero, BHK-21, and Tb1Lu cells, which were characterized by round shrinkage and exfoliation of cells, and these effects increased gradually over time, with 75% of the cells exhibiting CPE (+++ level) at 40–48 h (Table 1, Fig. 1). No obvious CPE was observed in C6/36 cells for 5 consecutive days in three successive passages after inoculation of both strains (Table 1, Fig. 1).
Time when the CPE reached the +++ level.
CPE, cytopathic effect; hpi, hour post infection.
To detect whether this virus can replicate in C6/36 cells, PCR was used to detect the S genes of WDBC1403 and WDBC1210 in the supernatants of the first, second, and third passages of C6/36 cells. The results were positive for all three passages, suggesting that although the strains WDBC1403 and WDBC1210 do not induce CPE, their viral genes can be replicated in C6/36 cells.
Macroscopic plaques were observed in BHK-21 cells after inoculation with WDBC1403. The plaques exhibited an irregular morphology with a diameter of 1.01 ± 0.28 mm (n = 8) and unclear margins (Fig. 2B). BHK-21 cells were inoculated with the strains WDBC1210 and WDBC1403 at an MOI of 0.1 for continuous culture. The titer of the WDBC1403 strain increased rapidly to a maximum of 1 × 106.57 pfu/mL at 24 h after inoculation, then gradually decreased to a minimum of 1 × 105.08 pfu/mL at 72 h. The maximum and minimum titers of the WDBC1210 strain were 1 × 108.33 and 1 × 107.41 pfu/mL at 24 and 72 h, respectively (Fig. 2C).
Whole-genome molecular features of the WDBC1403 strain
After the culture and harvest of BHK-21 cells infected with the WDBC1403 strain, the universal primers for flavivirus, alphaviruses, and bunyaviruses were used for PCR detection (Wang et al. 2011), and the results were negative. When KKV primers were used for PCR detection, the results were positive. Genome walking and RACE methods were used to obtain the whole genome sequences of the S, M, and L segments. PCR amplification primers (Supplementary Table S1) were used to confirm the complete nucleotide sequences of the WDBC1403 isolate and molecular characteristics were analyzed.
Viral genome sequence features
The S segment, which encodes the nucleocapsid protein (N; 233 amino acids) and the nonstructural protein (106 amino acids), is 982 nucleotides. The M segment, which encodes the glycoprotein precursor (GPC; 1414 amino acids), is 4515 nucleotides. The L segment, which encodes the RNA-dependent RNA polymerase (2249 amino acids), is 6891 nucleotides. The GenBank accession numbers of the S, M, and L segments of the WDBC1403 strain are MG014357, MG014358, and MG014359, respectively.
Comparison of the ORFs of WDBC1403 and KKV
When the nucleotide and amino acid sequences of the ORFs of the three genome segments of WDBC1403 were compared with the previously reported KKV PSC-19 (Groseth et al. 2014) and WDBC1210 isolates (Feng et al. 2017), the ORFs of the S and L segments were determined to be of equal length in the three strains. Two isoleucines were missing at the C-terminus of the GPC ORF of the M segment in WDBC1403 compared with the other two isolates. The nucleotide identities of the S, M, and L segments of WDBC1403 were 87.6%, 77.1%, and 78.3% with WDBC1210, and 88.2%, 76.7%, and 78.3% with PSC-19, respectively, whereas the amino acid identities were 96.6%, 85.9%, and 88.8% with WDBC1210, and 96.1%, 85.0%, and 88.9% with PSC-19, respectively (Table 2).
N
nt and aa similarity with WDBC1403.
nt length only includes the ORF, without the 3′- and 5′-UTRs.
Location in the corresponding ORF.
aa, amino acid; GPC, glycoprotein precursor; KKV, Kaeng Khoi virus; N, nucleocapsid protein; nt, nucleotide; NSs, nonstructural protein; ORF, open reading frame; RdRp, RNA-dependent RNA polymerase; UTRs, untranslated regions.
Characteristics of the 5′- and 3′-UTRs of WDBC1403
The nucleotide sequences of the 5′- and 3′-UTRs of the S, M, and L genome segments in WDBC1403 were obtained with 5′ and 3′ RACE. Both the 5′ and 3′ ends contain the 10-nucleotide reverse complementary sequences (5′-AGTAGTGTAC……GCACACTACT-3′) (Table 3; Supplementary Fig. S1) and were consistent with the conserved terminal sequence of the PSC-19 isolate.
In the WDBC1403 strain, the 5′-UTR lengths of the S, M, and L genome segments were determined to be 37, 23, and 26 nucleotides, respectively. The corresponding sequences in the PSC-19 and WDBC1210 isolates were nearly identical, with only three nucleotides in the S segment of WDBC1403 different from the other two strains (Supplementary Fig. S1).
The 3′-UTR lengths of the S, M, and L genome segments were determined to be 243, 247, and 115 bp, respectively, in the WDBC1403 strain. The S and L segments were 6 and 25 bp longer, respectively, and the M segment was 47 bp shorter, than the PSC-19 and WDBC1210 isolates. Overall, there were significant differences in the sequence of the WDBC1403 strain compared with the other two isolates, except for the conserved terminal sequence, whereas the sequences of the WDBC1210 and PSC-19 isolates were relatively similar (Supplementary Fig. S1).
Phylogenetic relationships of WDBC1403
To define the molecular genetic evolution of the WDBC1403 strain, the nucleotide sequences of the three genome segments of WDBC1403 were used to construct a molecular genetic evolutionary tree with the nucleotide sequences of representative strains of orthobunyaviruses (Fig. 3). Our analysis revealed that, regardless of the S, M, and L genome segments, the WDBC1403, PSC-19, and WDBC1210 isolates formed a new cluster in the Orthobunyavirus group. These strains also formed two evolutionary branches; the PSC-19 and WDBC1210 isolates belonged to one branch, whereas WDBC1403 formed an independent branch (Fig. 3). Molecular genetic evolution analysis of the sequence of the M segment showed that several KKV strains isolated from Cambodia in 2001 belong to the same evolutionary branch of the PSC-19 strain, which is in contrast with WDBC1403 (Fig. 3, M*).

Phylogenetic analysis of the WDBC1403 strain. Based on the whole-genome nucleotide sequences of the S, M, and L segments, partial nucleotide sequences of the M segment (510 nucleotides, M*) were used to construct the maximum-likelihood evolutionary tree. Bootstrap values based on 1000 replicates are also indicated. The WDBC1403 strain in this study is labeled with a black triangle. GenBank accession numbers: California Encephalitis virus (U12800 and AF123483), Inkoo virus (U47137, U88059, and EU789573), La Crosse virus (NC_004110, NC_004109, and NC_004108), Snowshoe hare virus (EU294510, EU262553, and EU203678), Trivittatus virus (KR149247, AF123491, and KR149249), Leanyer virus (HM627177, HM627176, and HM627178), Akabane virus (NC_009896, NC_009895, and NC_009894), Oropouche virus (NC_005777, NC_005775, and NC_005776), Batai virus (JX846595, JX846596, and JX846597), Bunyamwera virus (NC_001927, NC_001926, and NC_001925), Anhembi virus (JN572064, JN572063, and JN572062), Guaroa virus (KM245522, KM245523, and KM245524), Pongola virus (KJ867176, KJ867177, and KJ867178), Bwamba virus (KJ867182, KJ867183, and KJ867184), Nyando virus (KJ867188, KJ867189, KJ867190, KJ867194, KJ867195, and KJ867196), Mojui dos Campos virus (KJ867200, KJ867201, and KJ867202), and Kaeng Khoi virus (KJ867203, KJ867204, KJ867205, MF170064, MF170065, and MF170066).
Discussion
The original strain of KKV, PSC-19, was isolated from bats in Thailand in 1969 (Williams et al. 1976, Groseth et al. 2014). After this, 10 KKV strains were isolated from bat specimens collected in Cambodia in 2001. The nucleotide sequence differences of the partial M segment between these 10 strains and PSC-19 were only 2.6–3.2% (Osborne et al. 2003). In 2012, our laboratory isolated the WDBC1210 strain from bat flies collected at the China–Myanmar border. The nucleotide sequence of the M segment only differed by 5.9% with PSC-19 strain (Feng et al. 2017). It can be seen that, although the distances between Thailand and Cambodia, and between Thailand and the China–Myanmar border region, are ∼550 and 1400 km, respectively, and the difference in isolation time is decades apart (1969–2012), the genomes of KKV strains isolated in various regions are highly homologous and do not undergo major mutations. This suggests that the nucleotide and amino acid sequences of the KKV genome are relatively stable (Osborne et al. 2003, Feng et al. 2017).
However, the WDBC1403 strain isolated in the China–Myanmar border region, where WDBC1210 was previously isolated, exhibits obvious difference in the nucleotide and amino acid sequence of the S, M, and L segments. Especially the nucleotide difference of the M gene segment of WDBC1403 was 23%, the GPC ORF of the M segment lacked two amino acids, and the noncoding regions exhibited large differences, which may affect the antigenicity and pathogenicity of the virus.
Contrasting research found that the plaque diameter is smaller and the viral titer is approximately two orders of magnitude lower in the WDBC1403 strain than in the WDBC1210 strain (Table 1). The M segment in Bunyavirus encodes the membrane proteins, which is important for contact and fusion of the virus with cells (Hacker and Hardy 1997, Schmaljohn and Nichol 2007, Plyusnin et al. 2012), whereas differences in the viral UTRs affect viral replication titer (Barr and Wertz 2005, Mazel-Sanchez and Elliott 2012). Whether these phenotypic differences were caused by the genomic differences of the two strains, especially the ORF and UTRs of the M segment, still requires further investigation.
Although having obvious genome sequence difference with other KKV strains, according to the criterion of nucleocapsid protein amino acid, differences of more than 10% represent different species (Plyusnin et al. 2012); WDBC1403 whose nucleocapsid protein amino acid sequence differs by 3.9% with that of the original KKV, should be considered as a strain of KKV. Certainly, this needs to be further investigated by evaluating serological cross-reactivity to closely related viruses.
Groseth et al. (2014) showed that KKV belongs to the same evolutionary group as the mosquito-borne Nyando virus, and can replicate in C6/36 cells. In this study, the viral gene of KKV isolates was detected in the supernatant of C6/36 cells after three successive passages, which also suggests that KKV can replicate in C6/36 cells. These results suggest that the mosquito may be a potential transmission vector of KKV. Whether the mosquito can transmit KKV still needs to be validated by further research, which will likely include artificial infection experiments and virus isolation from field mosquito specimens.
Bat flies are blood-sucking parasites on the surface of bats (Tortosa et al. 2013). Therefore, they can carry and spread pathogens. Currently, Bartonella spp. has been detected in bat fly specimens ((Billeter et al. 2012, Morse et al. 2012, Brook et al. 2015). Orthoreovirus, orthobunyavirus, and rhabdovirus have been isolated or detected in bat flies in recent years (Jansen van Vuren et al. 2016, 2017, Goldberg et al. 2017). KKV was also isolated from bat fly specimens twice by our team. It has been suggested that bat flies may be a reservoir host or carrier for a variety of pathogens. In addition, neutralizing antibody to KKV was detected in staff members working in the cave where KKV was isolated in Thailand (Karabatsos 1985), suggesting that KKV is infectious in humans.
The WDBC1403 strain isolated in this study was shown to be a highly divergent KKV strain in terms of molecular biological characteristics, especially in the M segment, compared with previously isolated KKV strains. Whether these differences increase the pathogenicity of WDBC1403 isolated from bat flies to humans and animals warrants further study.
Footnotes
Acknowledgments
This work was supported by grants from National Natural Science Foundation of China (grant nos. 81290342 and 31560049), Yunnan Province Applied Basic Research Projects (grant no. 2016FB029), and Development Grant of State Key Laboratory for Infectious Disease Prevention and Control (grant no. 2014SKLID103), the National Key Research and Development Program of China (grant no. 2017YFC1200202), and the National Key R&D Program of China (grant no. 2016YFC1201904).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.