
Abstract
Coronaviruses (CoVs) represent a global public health threat, exemplified by the severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS) outbreaks. Using fecal samples collected from five bat species between 2014 and 2016 in Finland and RT-PCR, RT-qPCR, and NGS, we identified CoVs in 10 of 79 (13%) samples, including two novel bat species–CoV relationships. Phylogenetic analysis revealed Alphacoronavirus and Betacoronavirus species clustered among previously identified bat and human viruses. These results expand the known northern distribution and host species range of bat-borne CoVs.
Introduction
The emergence of severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS) established coronaviruses (CoVs) as a major global public health threat (Drexler et al. 2014). Spillover from wildlife sources is implicated for these emergence events, with subsequent research emphasis placed on documenting the wildlife host range and geographical distribution of CoVs to better understand the origins of human disease, and help design spillover mitigation strategies and improve public health readiness.
CoVs are classified into four genera, designated Alpha-, Beta-, Gamma-, and Deltacoronavirus (Woo et al. 2012, Drexler et al. 2014). Gamma- and Deltacoronavirus are predominately found in birds, whereas Alpha- and Betacoronavirus infect mammals, including humans (Woo et al. 2012). An especially high diversity of Alpha- and Betacoronavirus has been identified in bat species, supporting the relatively recent recognition of bats as major wildlife reservoirs for zoonotic viruses (Calisher et al. 2006) and indicating their ancestral host role in CoV evolution (Drexler et al. 2014). Despite this, minimal research has been conducted in northern ecosystems, including Fennoscandia (Finland and Scandinavia), where CoVs of bats have not been previously reported.
Materials and Methods
Noninvasive cross-sectional surveys were conducted in 17 separate sites in southcentral Finland between 2014 and 2016. Bats were captured using mist nets, harp traps, or by hand directly from roost sites, and individual fecal samples were collected from five bat species (Eptesicus nilssonii, Myotis brandtii, Myotis daubentonii, Pipistrellus nathusii, and Plecotus auritus). Bats were kept in cotton bags for up to 20 min and fecal pellets were collected from the bag or during examination. Bat species were identified using morphology, which is a sufficient method in Finland with only 13 known bat species. In addition to individual sampling, guano samples were collected from known roosting sites.
Samples were stored in RNAlater (QIAGEN, Hilden, Germany) in the field, and processed in two different laboratories using slightly different methods. RNA was extracted from the 2013 and 2014 samples using the QIAamp Viral RNA Mini Kit (QIAGEN) and primary screening was performed using the OneStep RT-PCR Kit (QIAGEN). We used previously described primers targeting the CoV RdRP-gene (11-FW 5′ TGATGATGSNGTTGTNTGYTAYAA 3′ and 13-RV GCATWGTRTGYTGNGARCARAATTC 3′; Muradrasoli et al. 2009). Positive samples were confirmed using the same RT-PCR kit and different previously described RT-PCR primers with slight modifications (Deltacoronavirus conserved primers, 5′ GGTTGGGACTATCCTAAGTGTG 3′, and 5′ CATCATCAGATAGAATCATCAT 3′; Woo et al. 2006), which yields a 440 bp amplicon.
After the project moved to a different laboratory with different reagents and established methods in 2015, RNA was extracted from the 2015 and 2016 samples using TriPure (Roche, Basel, Switzerland) and screened using the qScript One-Step SYBR Green qRT-PCR Kit, Low ROX (Quanta Biosciences, Beverly, MA) based on the same primers used for the 2013 and 2014 samples (same 11-FW as mentioned and slightly modified 13-RV GCATWGTRTGYTGNGARCARAATT 3′, no probes; Muradrasoli et al. 2009). Positive samples were reverse transcribed to cDNA using the RevertAid Premium Kit (ThermoFisher Scientific, Waltham, MA) and confirmed through a previously described nested RT-PCR method (de Souza Luna et al. 2007), which yields ∼400 bp amplicon on the nested round, using a Phusion Flash High-Fidelity PCR Master mix (ThermoFisher Scientific). PCR amplicons were sequenced using Sanger sequencing.
Extracted RNA from CoV-positive samples was also used for sequencing on a MiSeq platform (Illumina, San Diego, CA). Preparation of the samples and assemblies of the reads was done as described previously (Forbes et al. 2019). Quality of the assemblies was assessed by aligning reads to contigs using BWA alignment algorithm in UGENE (1.29.0 64-bit version; Okonechnikov et al. 2012). The same method with reference sequences instead of contigs was used to retrieve cytochrome b and cytochrome c oxidase subunit I sequences from MiSeq reads of guano samples for host species identification (available upon request).
Sequences acquired from Sanger and MiSeq sequencing were aligned (Muscle 3.8.31) with sequences of CoV RdRP from GenBank, after which the best substitution model was selected (MEGA7 7.0.26 Model Selection [ML] feature with default settings). A phylogenetic tree was constructed using a Bayesian framework with MrBayes (3.2.6 × 64; Ronquist et al. 2012).
Results
Ten of 79 total samples contained CoV RNA (13%) and at least 1 positive sample was obtained from three of the five different bats species, with CoV prevalence ranging from 6% to 33% (Table 1). The exceptions were P. nathusii and P. auritus.
Total Number of Each Sample Type Collected from Each Bat Species in Finland (Number of Positive Samples/Total Number of Samples)
CoV sequences were obtained from nine samples: two from M. daubentonii roost sites (both alpha-CoV, GenBank accession numbers MG923567.2 [776 bp] and MG923569.1 [395 bp]), four from M. daubentonii (all alpha-CoV, GenBank accession numbers MG923568.2 [622 bp], MG923571.2 [4805 bp], MG923573.2 [15 837 bp], and MG923574.2 [28 045 bp]), two new CoV–host relationships; M. brandtii (two alpha-CoVs, GenBank accession numbers MG923572.2 [436 bp], and MN065811.1 [28 119 bp]) and E. nilssonii (beta-CoV, GenBank accession number MG923570.1 [395 bp]). The constructed phylogenetic tree showed sequences belonging to Alphacoronavirus and Betacoronavirus genera, which were clustered among known bat and human viruses (Fig. 1).
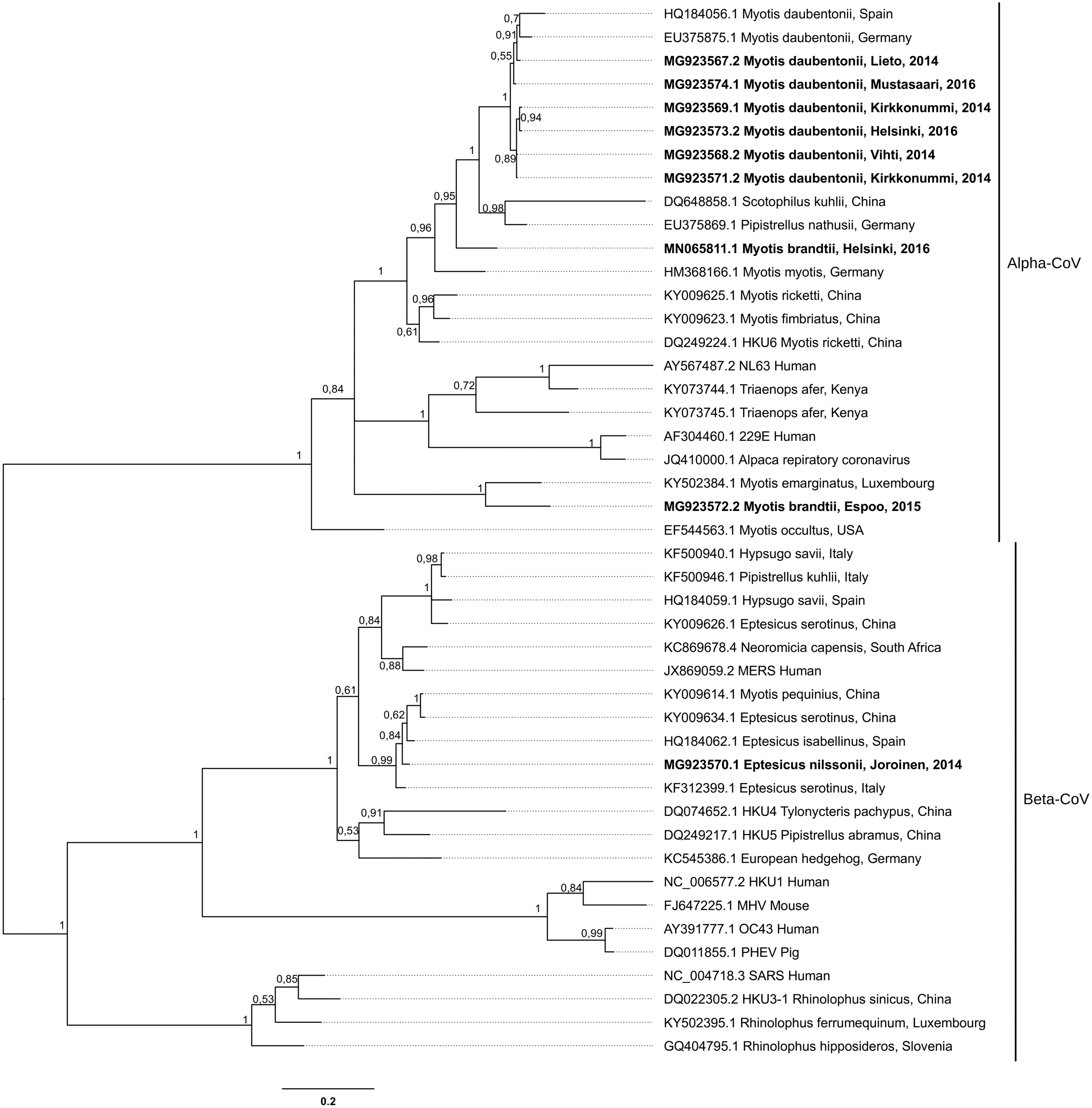
Phylogenetic consensus tree indicating the evolutionary relationships among partial coding sequences of the CoV RdRP-gene (392 nucleotides) from Finnish bats and CoVs found elsewhere in bats and other species. Terminal node information includes the bat species, and the location and year of sample collection (year of only the samples reported in this study). Posterior probabilities of internal nodes are located next to given nodes and the scale bar shows expected nucleotide changes per site. Bold face nodes represent sequences reported in this study. The tree was made using MrBayes 3.2.7a × 64 with a GTR + G nucleotide substitution model, two Markov chain Monte Carlo (MCMC) runs with four chains, 1,000,000 samples, a sampling frequency of 1000, and a burn-in of 25%. CoV, coronavirus.
Discussion
This report increases knowledge on the geographical distribution and host species range of bat-borne CoVs. We show for the first time that bat-borne CoVs occur in northern Europe, and identified two previously unknown bat species–CoV associations.
The alpha-CoV sequences from M. daubentonii, one of which was from a bat captured in Mustasaari (GenBank accession number MG923574.2) on the west coast of Finland and another from a guano sample collected from bat colony in Lieto (MG923567.2) in the southeast corner of Finland, clustered among sequences from central and southern Europe, supporting previous findings of gene flow among European populations of this host (Laine et al. 2013). Other CoV sequences from M. daubentonii were from the southern coast and formed their own cluster.
Meanwhile, one of the alpha-CoVs from M. brandtii (MN065811.1) clustered among other CoVs of Myotis spp. from Europe. There is a possibility that this is due to a cross-species transmission, since another alpha-CoV from M. brandtii (MG923572.2) was phylogenetically separated from those sequences, with one exception being a sequence from M. emarginatus from Luxembourg, which parallels phylogenetic reconstructions made of the Myotis genus (Ruedi et al. 2013). The Beta-CoV from E. nilssonii clustered with Betacoronaviruses of other Eptesicus spp. Most of these findings indicate host fidelity and the codiversification of CoVs with their host species.
Although we show that bat species in northern Europe are host to CoVs, potential human health concerns are premature based on these initial findings, and must be balanced against the important environmental services that bat species provide. Bats play a critical role in ecosystem functioning, including insect pest management, seed dispersal, and pollination (Kunz et al. 2011), and are protected in Finland and most other parts of Europe.
Our identification of bat-borne CoVs in northern Europe, including two novel host–CoVs relationships, will enable more in-depth study about the pathogens carried by bats living in urban, rural, and forest habitats in northern latitudes, their evolutionary relationships with host species, and potential risks to human health. Further research, including whole genome targeting methods (e.g., NGS) from fresh samples from wider geographical and temporal ranges, is required to better understand the evolutionary history and epidemiological properties of these CoV–host relationships, and ultimately the risks they may pose to human health.
Footnotes
Acknowledgments
We are very grateful to Emma Hakanen, Petra Rinne, Merja Hautala, John Loehr, Anne Jääskeläinen, Mira Utriainen, and Teemu Smura for their field, laboratory, and NGS data analysis assistance.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
Funding for this study was provided by the Jenny and Antti Wihuri Foundation, the Finnish Foundation of Veterinary Research, the Finnish Biological Society Vanamo, the Oskari and Siiri Vilamo Fund, and the Häme Students Foundation. The research was conducted under permits from the Centres for Economic Development, Transport, and the Environment (permits POSELY/501/07.01/2010, UUDELY/475/07.01/2011, VARELY/433/07.01/2011, EPOELY/2239 and 2292/2014, KASELY/1022/2014, POKELY/585/07.01/2014, UUDELY/3001/2014, VARELY/948/2014, and HAMELY/1190/2015).