
Abstract
Ticks are important vectors for different tick-borne viruses, some of which cause diseases and death in humans, livestock, and wild animals. Tick-borne encephalitis virus, Crimean-Congo hemorrhagic fever virus, Kyasanur forest disease virus, severe fever with thrombocytopenia syndrome virus, Heartland virus, African swine fever virus, Nairobi sheep disease virus, and Louping ill virus are just a few examples of important tick-borne viruses. The majority of tick-borne viruses have RNA genomes that routinely undergo rapid genetic modifications such as point mutations during their replication. These genomic changes can influence the spread of viruses to new habitats and hosts and lead to the emergence of novel viruses that can pose a threat to public health. Therefore, investigation of the viruses circulating in ticks is important to understand their diversity, host and vector range, and evolutionary history, as well as to predict new emerging pathogens. The choice of detection method is important, as most methods detect only those viruses that have been previously well described. On the other hand, viral metagenomics is a useful tool to simultaneously identify all the viruses present in a sample, including novel variants of already known viruses or completely new viruses. This review describes tick-borne viruses, their historical background of emergence, and their reemergence in nature, and the use of viral metagenomics for viral discovery and studies of viral evolution.
Introduction
The rate of emergence and reemergence of infectious diseases is increasing worldwide and presents a significant global economic burden. A significant proportion of these diseases is dominated by zoonoses that originate from wildlife. During recent years a number of pathogen crossovers from wildlife to humans and livestock have occurred that resulted in emerging disease threats. East African fever caused by Theileriaparva in cattle (Blancou 2005), Hantavirus Pulmonary Syndrome, Nipah virus disease, and Hendra virus disease originated from wildlife sources (Delwart 2000).
Ticks are important vectors and reservoirs of a broad range of pathogens that are capable of causing diseases in humans, livestock, and wild animals (Estrada-Peña et al. 2013). To date, at least 38 known viral species are transmitted by ticks, and some are significant threats to human and animal health (Estrada-Peña et al. 2014). Such viruses include tick-borne encephalitis (Katargina et al. 2013, Puchhammer-Stockl et al. 1995, Skarpaas et al. 2006, Crimean-Congo hemorrhagic fever virus, Kyasanur forest diseases virus (KFDV), severe fever with thrombocytopenia syndrome virus (SFTSV), Heartland virus (HRTV), African swine fever virus (ASFV), Nairobi sheep disease virus, and Louping ill virus (Estrada-Peña et al. 2014).
The interaction of domestic animals, humans, and wildlife can promote tick-borne spread of zoonotic pathogens. Nomadic and pastoralist lifestyles, especially those at the wildlife-livestock interface, can allow direct and indirect contact, which can facilitate exposure and sharing of previously isolated pathogens (Labuda et al. 2004).
Since many viruses cannot be cultivated and lack common viral gene markers, detection of viruses using conventional, immunological, and PCR methods is very difficult (Bexfield and Kellam 2011). Furthermore, many as yet undiscovered viruses cannot be identified using these specific methods. This review focuses on tick-borne viruses and how viral metagenomics represents a powerful tool to investigate emerging and reemerging tick-borne viruses.
Tick Characteristics
Ticks are one of the most important arthropod vectors for wild and domestic animals as they harbor a wide range of pathogens such as viruses, bacteria, and protozoa. Ticks are obligate ectoparasites of mammals, birds, and reptiles, and around 900 species of ticks are known (Estrada-Peña et al. 2013). As part of the subphylum Chelicerata, class Arachnida, subclass Acari, and suborder Ixodida, ticks can be classified into three recognized families: Ixodidae (hard ticks, 692 species), Argasidae (soft ticks, 186 species), and Nuttalliellidae (1 species) (Walker 2003). All hard ticks have a sclerotized dorsal plate that is absent in soft ticks. Adults have dorsoventral flattening and are oval in shape (Estrada-Peña et al. 2014). The main tick species that transmit pathogens of veterinary and medical importance and affect domestic and wild animals are principally found in the hard tick genus of Rhipicephalus, Amblyomma, Hyalomma, and Haemaphysalis (Walker 2003). In addition to pathogen transmission, some tick species can also harm their host through injection of toxins to induce paralysis (Estrada-Peña et al. 2014).
Unlike hard ticks, the Argasidae (soft ticks) lack hard plates, called scutum, on their dorsal surface. Species of this genus have a worldwide distribution. They also display dorsoventral flattening and have a general oval outline, although their shape varies between species. Medically important soft-tick species belong to the genus Ornithodoros (Walker 2003). The development of soft ticks differs in several ways, in that, the majority of the species requires multiple hosts to complete their life cycle (Estrada-Peña et al. 2013).
Factors for Emerging Tick-Borne Viruses
Given that ticks are important vectors of recent emerging zoonotic infections (Piantadosi et al. 2016) an understanding of how they influence the evolution of their associated pathogens that might lead to outbreaks is critical. Emerging infectious disease (EID) outbreaks may involve biological expansion due to population increase or geographic spread of parasites and pathogens from their original areas into new areas or ecological changes, resulting in phenological shifts among species (Brooks et al. 2006).
When hosts and their associated pathogens move outside their areas of origin and ecosystems, host-switching may occur in another phylogenetically conserved host-parasite interaction, through a process known as ecological fitting (Brooks et al. 2006). Recent studies have continued to highlight the increased possibilities of host-switching in ticks based on observations of zoonotic tick-borne pathogens in unlikely hosts apart from their normal reservoir hosts (Estrada-Peña et al. 2014).
Increasing anthropogenic activities have largely contributed to the current trends of EIDs (Lindahl and Grace 2015). Biodiversity loss, which is intrinsically related to climate change, has been accelerated by human activities. Such changes have altered tick population structures as well as their boundaries in recent years. Notably, the consequences of biodiversity loss involve not only species extinction but also a crisis of introduced species and emerging diseases (Brooks et al. 2006). Frequently identified factors that contribute to disease emergence include ecological changes involving those due to agricultural or economic development, human demographics and behavior, international travel and commerce, technology and industry, microbial adaptation, and changing ecosystems (Morens et al. 2012).
Historical Background and Characteristics of Emerging and Reemerging Tick-Borne Viruses
Crimean Congo hemorrhagic fever virus (CCHFV) was first described during an outbreak of hemorrhagic fever in Red Army soldiers in the Crimea (Mertens et al. 2013). The virus was shown to be antigenically similar to a second virus isolated from humans in the Democratic Republic of Congo (Mertens et al. 2013). CCHFV is primarily transmitted by ticks of the genus Hyalomma and has one of the most extensive distributions of any tick-borne virus, with infections reported in the Middle East, Asia, and Sub-Saharan Africa (Mertens et al. 2013). CCHFV belongs to the order Bunyavirales, family Nairoviridae, and genus Orthonairovirus, and is characterized by a trisegmented negative-sense RNA genome that encodes four structural proteins, the nucleoproteins, glycoprotein, and RNA-dependent RNA polymerase (Table 1). The virus is particularly virulent in humans, but has little or no pathogenic effect on livestock (Mertens et al. 2013).
Summary of Emerging and Reemerging Tick-Borne Viruses
Early cases of SFTSV were reported between 2007 and 2010 in Henan and Hubei provinces in China (Lam et al. 2013). Investigation of the first outbreak revealed that patients with SFTSV infection were farmers with a history of tick bites. SFTSV is a bunyavirus related to viruses in genus Phlebovirus that is characterized by a single-stranded negative-sense genome having three segments coding for the nucleoproteins, surface glycoprotein, and RNA-dependent RNA polymerase (Table 1) (Liu et al. 2014). This virus has not been reported in livestock, suggesting that humans are particularly susceptible to SFTSV infection. These viruses are distributed mainly in East Asia and North America where Haemaphysalis longicornis serves as the primary tick vector (Liu et al. 2014).
HRTV was reported for the first time in 2009 in North America when two farmers from Missouri were admitted to the hospital (McMullan et al. 2012). HRTV was identified in Amblyomma americanum, an abundant tick species in many regions of the United States (Table 1) (McMullan et al. 2012). However, HRTV exists in different tick species and has emerged on different continents. HRTV belongs to the order Bunyavirales, family Phenuiviridae, and genus Phlebovirus, characterized by a trisegmented negative-sense RNA strand genome (McMullan et al. 2012).
KFDV was first reported in 1957 following an outbreak of hemorrhagic fever among villagers living in the Kyasanur forest area in India (Holbrook 2012). KFDV is a member of the genus Flavivirus, family Flaviviridae characterized by a positive-sense single-stranded RNA genome that codes for three structural and seven nonstructural proteins (Table 1) (Holbrook 2012). The virus is mainly distributed in India and the Haemaphysalis spinigera is the primary tick vector (Holbrook 2012).
Alkhurma hemorrhagic fever virus (AHFV) is a recently described tick-borne virus that was first isolated in 1995 in Alkhuma city, Saudi Arabia (Horton et al. 2016). AHFV belongs to the family Flaviviridae, genus Flavivirus and is characterized by a positive-sense RNA strand genome (Table 1) (Horton et al. 2016). AHFV is mainly distributed in Saudi Arabia and it has been identified in both the Argasid tick Ornithodoros savignyi and Ixodid tick Hyalomma dromedary (Horton et al. 2016).
Deer tick virus (DTV) was originally isolated from the Rocky Mountain wood tick, Dermacentor andersoni, but is mainly found in Ixodes scapularis (Aliota et al. 2014). A member of the family Flaviviridae, genus Flavivirus, DTV is characterized by a positive-sense RNA strand genome (Table 1) (Aliota et al. 2014). The virus is distributed mainly in North America where I. scapularis is the primary tick vector (Aliota et al. 2014).
ASFV is the causative agent of an acute hemorrhagic fever that causes severe morbidity and high mortality in domestic pigs (Mur et al. 2016). ASFV is classified within the family Asfarviridae, genus Asfivirus,and consists of a double-stranded DNA genome that encodes over 150 proteins (Table 1). The genome is surrounded by a protein capsid and a host-derived envelope (Mur et al. 2016). ASF was first described in Kenya and is found across Sub-Saharan Africa, Asia, Europe, and China (Mur et al. 2016). The sylvatic cycle occurs in bush pigs (Potamochoerus larvatus) and warthogs (Phacochoerus africanus) that are infected following infestation with Argasid ticks of genus Ornithodoros, particularly Ornithodoros moubata (Thomson 1985).
Viral Metagenomics to Detect Tick-Borne Virus
Many microorganisms cannot be cultivated, and thus often go unnoticed due to difficulties in study using cell culture methods (Bexfield and Kellam 2011). Likewise, for viruses, various molecular methods rely on known markers that have been used to detect specific viruses. Viral metagenomics involves the study of all genetic material extracted from a sample for pathogen detection, virus discovery, and outbreak preparedness, and does not require viral culture, marker genes, or any previous knowledge of what pathogens might be present (Bexfield and Kellam 2011).
Recently, metagenomics has been used to study viral populations in a number of environmental samples as well as different host and vectors. Metagenomics profile analysis of the viral populations carried in Rhipicephalus spp. ticks found in China identified some sequences originating from viruses that had not been described previously in ticks (Xia et al. 2015). A viral metagenomics study by Cholleti et al. (2018) revealed the presence of highly divergent quaranjavirus in Rhipicephalus ticks from Mozambique. Souza et al. (2018) examined the viral diversity of Rhipicephalus microplus and found several new variants from previously known viral families such as Phenuiviridae, Chuviridae, and Flaviviridae. Because these techniques investigate complete viral communities within a sample, both known and newly emerged viruses can be identified (Blomstrom 2011). The following sections describe major techniques used to study viral metagenomes associated with ticks.
Basic Approaches to Tick-Borne Viral Metagenomics
Collection of ticks
Ticks can be collected from the body of domestic animals and wild animals, as well as from the environment (free living, also termed questing ticks). During visual examination of ticks, attention should be given to the abdomen, back, anal area, and legs (Estrada-Peña et al. 2014). Ticks collected from animals should be removed manually and placed in sterile plastic vials for transport to the laboratory.
Domestic animals can be physically restrained for tick collection, whereas for collection of ticks from wild animals, wildlife service veterinarians should immobilize the animal using a combination of etorphine hydrochloride and xylazine hydrochloride (Estrada-Peña et al. 2013).
Questing (free living) ticks are usually collected from purposefully chosen areas within logistical constraints, and include host resting areas and burrows, host routes, and areas surrounding watering holes (Estrada-Peña et al. 2014). Collection methods for questing ticks include dragging a flag, hand picking from vegetation, and carbon dioxide trapping using improvised carbon dioxide traps (Walker 2003).
Following collection, the ticks are transported live to the laboratory in tubes plugged with cotton swabs. In the laboratory, the sampled ticks are washed with sterile water to remove excess environmental particulate contamination and then rinsed with 70% ethanol (Estrada-Peña et al. 2014). Minimum essential medium containing antimicrobial agents is sometimes used to protect ticks from microbes (Estrada-Peña et al. 2013). The washed ticks are then transferred to sterile vials and stored at −80°C until processing for identification and virus isolation (Estrada-Peña et al. 2014).
Sample preparation and nucleic acid extraction
Because viral genomes often constitute a small proportion of the total nucleic acids from an extracted sample, sample preparation is the most important and challenging aspect of viral metagenomics (Cholleti et al. 2018). Pooling ticks is a common approach to increase the likelihood of virus identification (Blomstrom 2011). A major obstacle for virus detection in ticks using viral metagenomic approaches is contamination by both host and nonhost nucleic acids. Several approaches can be used or combined during sample preparation to deplete these potential contaminants, while retaining viral nucleic acids (Cholleti et al. 2018) (Fig. 1).
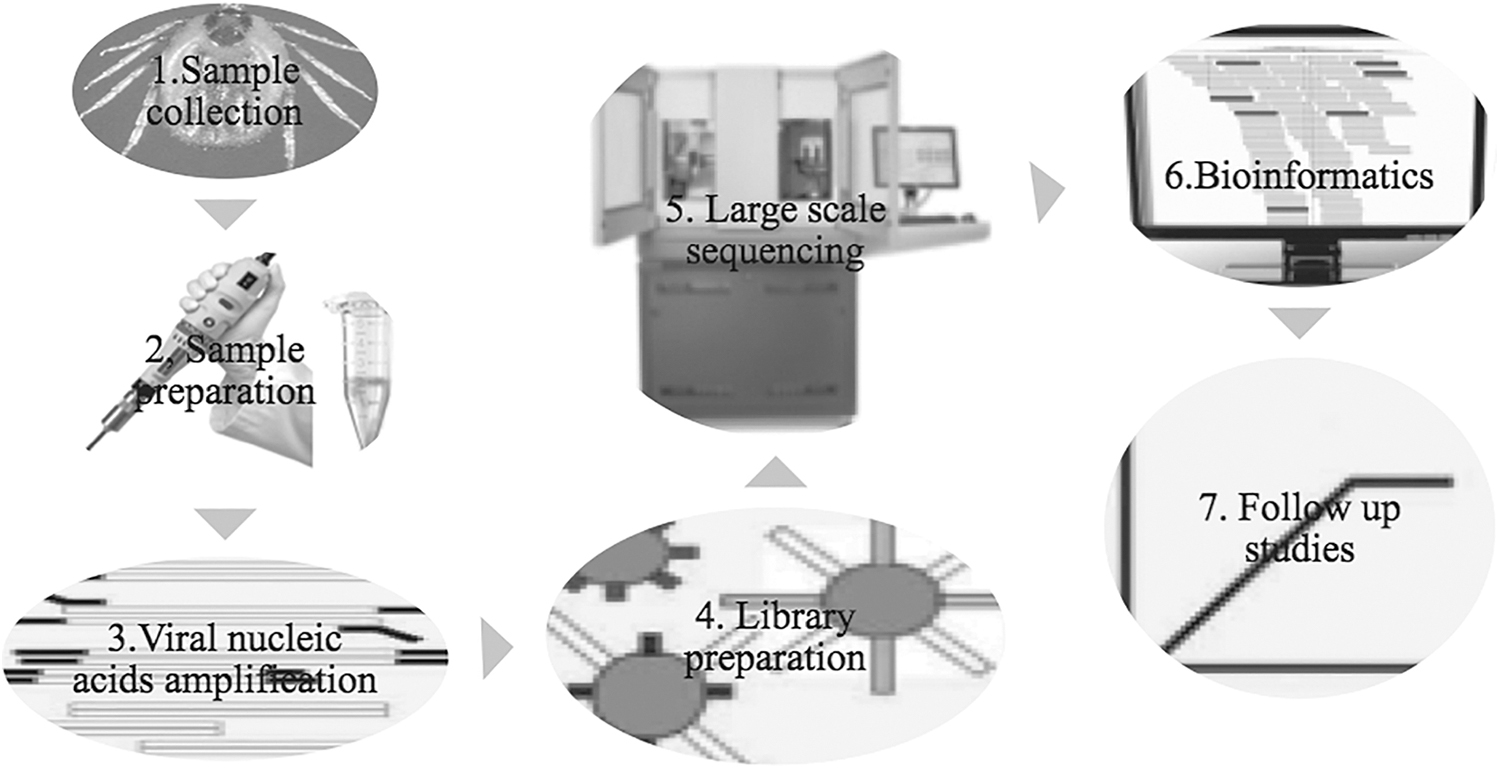
Diagram of viral metagenomics study work flow for discovery of viruses in tick samples using sequence-independent amplification and HTS. HTS, high-throughput sequencing.
Density centrifugation methods involving sucrose or cesium chloride gradients are widely used to separate the viral particles from other components. Filtration using a 0.22-μm filter is a common procedure for removing bacteria and other cells, while retaining smaller virus particles is also frequently used when searching for viruses (Cholleti et al. 2018). However, several viruses are as large as bacteria and can also be removed by filtration. For example, mimiviruses are DNA viruses that have an ∼1.2 Mb genome and a capsid diameter of ∼500 nm, and thus would likely be removed by 0.22-μm pore filters (Xia et al. 2015).
Treating samples with nucleases before nucleic acid extraction degrades host nucleic acids, while preserving virus nucleic acids, which are presumably protected by the viral capsid (Blomstrom 2011). Currently, DNase treatment is included in most viral metagenomics approaches (Cholleti et al. 2018). RNase treatment is frequently used to remove host RNA. However, a major problem associated with RNA contamination is that host- and bacteria-derived ribosomal RNA (rRNA) is difficult to degrade and a high proportion of rRNA often remains even after RNase treatment (Cholleti et al. 2018). Thus, many nuclease methods can reduce, but not completely eliminate, the content of host nucleic acids, particularly in tissue samples (Bexfield and Kellam 2011). Meanwhile, some viruses are more sensitive to nuclease treatment than others, increasing the likelihood that viral nucleic acids may also be degraded (Bexfield and Kellam 2011). Among non-nuclease approaches to eliminate rRNA, a set of 96 specific primers instead of random primers can be used during cDNA synthesis (Bexfield and Kellan 2011). These primers are designed to target all known viruses, but do not prime rRNA. Ribosomal sequences can also be extracted from samples. One example of this approach is Ribo-Zero technology that contains a number of highly conserved ribosomal sequences, including those from ticks, which allow hybridization between the synthetic sequences and those in the sample for subsequent removal of rRNA (Bexfield and Kellam 2011).
Viral nucleic acid amplification strategies
Viral metagenomics use sequence-independent amplification assays that require no specific primer or probe (Blomstrom et al. 2016). Amplification of the nucleic acids is performed in a sequence-independent manner to reveal the true genetic composition of the sample and allows simultaneous increases in the amounts of several viral genomes, including highly divergent and completely novel viruses that enable their discovery and genetic characterization (Xia et al. 2015). Various strategies for amplification have been used to randomly amplify all of the nucleic acids in a given sample (Blomstrom et al. 2016). Some of the more commonly used sequence-independent strategies are briefly described in the following sections.
Multiple displacement amplification
Multiple displacement amplification is an efficient amplification strategy that uses random primers combined with a displacement polymerase. Phi29 is a high-fidelity, highly processive displacement polymerase that can incorporate over 70,000 bases before detaching from a template (Blanco et al. 1989), and requires only a few nanograms of starting material to produce several micrograms of DNA in a single reaction. Using random primers and phi29, circular targets can be efficiently amplified through a reaction termed rolling-circle amplification, which creates multiple copies of the target (Dean et al. 2001). Circular targets are ideal for this amplification strategy, although linear targets can also be amplified in a similar manner (Berthet et al. 2008). However, amplification of nucleic acids from short linear DNA viruses and RNA viruses is less efficient, and additional steps, such as ligation, may be needed (Berthet et al. 2008). This strategy has been successfully used for the study of several different viral metagenomes, and multiple novel viruses have been discovered using this method (Berthet et al. 2008).
Sequence-independent single-primer amplification
Sequence-independent single-primer amplification (SISPA) is based on endonuclease restriction of target DNA followed by ligation of adaptor linkers that are complementary to overhanging bases on the target DNA (Cholleti et al. 2018). RNA viruses can also be amplified by SISPA following random-primed cDNA synthesis. In this approach, dsDNA is synthesized using the DNA polymerase activity of reverse transcriptase. RNAse H is used to digest the RNA component of the RNA/DNA hybrid (Cholleti et al. 2018). A PCR primer complementary to the ligated linker is then used to amplify the sequences located between pairs of restriction sites (Blomstrom 2011). Common end sequences of the adaptor allow the DNA to be amplified in a subsequent PCR using a complementary single primer.
Due to the low complexity of a viral genome, enzymatic digestion produces a large amount of a limited number of fragments (Cholleti et al. 2018). These methods have several advantages, including (1) an ability to detect novel viruses that are highly divergent from previously described viruses; (2) relative speed and simplicity; and (3) a lack of bias in identifying particular groups of viruses (Delwart 2007). Limitations of sequence-independent amplification techniques arise from the potential for amplification of contaminating host and bacterial nucleic acids (Blomstrom 2011).
Single-primer isothermal amplification
This amplification technology uses a single chimeric primer for the amplification of DNA (single-primer isothermal amplification [SPIA]) and RNA (Ribo-SPIA). SPIA employs a single, target-specific chimeric primer comprising deoxyribonucleotides at the 3′ end and ribonucleotides at the 5′ end, together with RNase H and a DNA polymerase having a strong strand displacement activity (Blomstrom et al. 2018). Amplification is initiated upon hybridization of the chimeric primer to a complementary sequence in the target DNA molecule.
The DNA polymerase then initiates primer extension of the hybridized primer along the target DNA strand (Myrmel et al. 2017). The RNaseH then mediates cleavage of the 5′ RNA portion of the extended primer (RNA-DNA hybrid), thus freeing part of the primer binding site on the target DNA strand to allow binding of a new chimeric primer (Blomstrom et al. 2018). The newly bound primer competes with the previous primer extension product for binding to the complementary DNA target sequence and is stabilized by binding of DNA polymerase that displaces the 5′ end of the previous extension product. As replication is again initiated by primer extension, RNase H cleavage of the 5′ RNA portion of the newly extended primer frees part of the primer binding site for subsequent primer binding and the replication cycle is repeated (Myrmel et al. 2017).
SPIA can be used for global genomic DNA amplification and for amplification of specific genomic sequences and synthetic oligonucleotide DNA targets. Ribo-SPIA is similarly suitable for global and target-specific RNA amplification (Blomstrom et al. 2018). Ribo-SPIA technology provides an elegant method for linear, isothermal amplification of mRNA species in a total RNA population (Myrmel et al. 2017). Therefore, this process can be used to amplify large populations of nucleic acid species, which are often limited in biological samples that are commonly encountered in clinical research. Moreover, this approach provides high coverage and frequently produces full-length genomes of viruses present in the samples (Myrmel et al. 2017).
Random PCR
Random amplification protocols avoid the need for restriction enzyme digestion and ligation. Random PCR is an alternative sequence-independent amplification technique that is commonly used to amplify known viruses and has also been used for the identification of novel viruses (Bexfield and Kellam 2011). This approach requires no adaptor ligation step and, compared with conventional PCR, which uses one pair of complementary forward and reverse primers to amplify DNA in both directions, random PCR involves two different primers and two separate PCRs (Blomstrom et al. 2016).
The single primer used in the first PCR has a defined 5′ sequence followed by a degenerate hexamer or heptamer sequence at the 3′ end. A second PCR is then performed with a specific primer complementary to the 5′ defined region of the first primer, thus enabling amplification of products formed in the first reaction (Blomstrom et al. 2016). Random PCR has been used extensively to detect both DNA and RNA viruses and is currently the molecular method that is most commonly used to identify unknown and novel viruses (Blomstrom et al. 2016). Although random priming can amplify unknown targets, this method often yields lower amounts of DNA than that produced using specific primers, which can reduce the overall sensitivity of the process.
Pooling strategy
High-throughput sequencing (HTS) technology has decreased the cost of sequencing tremendously in recent years (He et al. 2011). However, to increase the likelihood of virus identification using viral metagenomic studies, large sample sizes are needed (He et al. 2011). Unfortunately, sequencing of such large sample sizes remains expensive. Several steps contribute to the actual cost of sequencing a sample. First, the samples must be prepared, which involves treatment with nuclease and depletion of contaminating rRNA before amplification according to the selected method. These steps are often not factored into calculations of total sequencing costs. Another cost accompanies library preparation from DNA samples for sequencing. Finally, the cost of the actual sequencing is proportional to the amount of sequence to be characterized and is referred to as the sequencing per-base cost.
Technological advances are rapidly reducing the per-base cost of sequencing, whereas the cost for library preparation has remained stable (Erlich et al. 2009). To reduce library preparation costs, the concept of DNA pooling was introduced. The basic idea behind this approach is that DNA from multiple individuals is pooled into a single DNA mixture that is then prepared as a single library and sequenced (Erlich et al. 2009). This approach reduces library preparation costs because only one library is prepared per pool instead of one library per sample. Pooling has been successfully used to detect rare virus variants (Craig et al. 2009), which supports its use in viral metagenomics studies to detect known and unknown viruses (Xia et al. 2015, Cholleti et al. 2018).
However, pooling methods assume that all individuals in the pool have the same abundance of viral nucleic acids (Erlich et al. 2009). The abundance level for each individual is the fraction of the reads in a pool that originated from that specific individual (Erlich et al. 2009). However, this simple assumption is often false, given that some individuals can have different abundance levels, which produce spurious associations. As such, a thorough plan to optimize pooling strategies based on the scope of the study is highly important.
Sequencing Technologies
Most PCR methods generate products that require definitive identification. Sequencing is often used to identify viral nucleic acids in a given sample. Standard sequencing technologies such as Sanger sequencing (Sanger et al. 1977) produce high-quality sequence data and can currently produce sequence reads of up to 850 bp (Table 2). A limitation of this technique in terms of virus identification is the requirement for cloning viral sequences into bacterial vectors before sequencing, although direct sequencing of PCR products can also be used. When cloning is performed using this method, host-related bias can occur (Sanger et al. 1977). Since only relatively few clones can be sequenced, methods to enrich for virus before amplification are required. Such approaches are highly laborious compared to the yield. Therefore, new HTS technologies are the only choice for metagenomic studies. A combination of Sanger sequencing and advanced fluorescent detection methods, developed in association with the HUGO project, led to the development of HTS approaches that are often referred to as next-generation sequencing (Fig. 1).
Summary of Output Read Length, Sequencing Depth, and Bases Per Run for Currently Available Sequencing Technologies
High-throughput sequencing
The 454 platform by Roche was the first commercial HTS technology to be released and was followed by platforms from Illumina and the SOLiD system (Pareek et al. 2011). The different approaches to clonal amplification and sequencing strategies used for these three technologies translate to differences in the amount of data produced and the read length. The 454 and SOLiD systems utilize emulsion PCR for clonal amplification, whereas the Illumina system uses bridge PCR. The 454 platform is based on pyrosequencing and produces reads of ∼400 nucleotides with a 0.4–0.6 Gb/run throughput (Table 2). The bridging PCR used by the Illumina system involves reversible terminators and has a throughput of 3–6 Gb/run with a read length of ∼100 nucleotides. The SOLiD platform based on ligation and cleavable probes has a throughput of 10–20 Gb/run, but the read length is only ∼50 nucleotides (Pareek et al. 2011). The various benefits and drawbacks to these sequencing technologies should be considered when choosing between the different platforms (Table 2).
For viral metagenomics studies, the 454 platform was originally widely used because of its longer read lengths that facilitated de novo assembly. The 454 system is now no longer used for viral metagenomics and has been replaced by Illumina technologies. New platforms are also being introduced, including Ion Torrent (Life Technologies), which is based on the release of hydrogen ions as nucleotides are incorporated by the polymerase (Pareek et al. 2011).
Although the abovementioned sequencing technologies do not require bacterial cloning, each product must still be clonally amplified before sequencing. However, new single-molecule sequencing technologies that do not require this step are in development (Pareek et al. 2011). In 2007, PacificBiosciences released the first single-molecule sequencing platform (Harris et al. 2008). Meanwhile, Oxford Nanopore (MinION) sequencing involves threading of DNA through a nanopore and recording of each base as it moves through the pore, and represents another promising single-molecule technology under development, which could provide substantially longer read lengths. The throughput and read length of both the Pacific Biosciences and MinION are 5 million per run and >20,000 nucleotides, respectively (Pareek et al. 2011) (Table 2). Single-molecule sequencing can have advantages as this approach avoids errors that are introduced by the presequencing amplification and also provides a more faithful picture of the frequencies of different DNA fragments. Although HTS is always required for viral metagenomics screening, Sanger sequencing is still often used for follow-up studies due to its capacity to produce longer sequencing reads and fill missing gaps.
Bioinformatics
Bioinformatics is the application of tools and computational analyses to understand and interpret biological data. Bioinformatics is an interdisciplinary field that has been widely applied in modern biology and medicine for data management (Bayat 2002). The analysis of massive sequencing data generated from HTS platforms typically includes quality checking, assembly, and taxonomic classification of contigs produced by the assembly.
Quality control and trimming
Quality checking involves trimming of sequences according to Phred quality scores, which are related to base calling error probabilities (Ewing et al. 1998). Identifying and removing sequence duplicates produced by the HTS platform and that result from PCR amplification, PCR errors, or sequencing errors is another aspect of quality checking that is needed to reduce computational time, accurately calculate estimated species abundance, and improve assembly (Bokulich et al. 2013). All of these quality filtering conditions can be specified depending on the downstream analyses required (Bokulich et al. 2013). Quality checking can be performed using several tools such as PRINSEQ, FASTQC, Scythe, Sickle, and Fastp (Bokulich et al. 2013).
Filtering and alignment
Sequences that are not targets of study can be filtered out to eliminate misassemblies and to speed the analysis. For example, host sequences can be removed from a sample if the target sequences are viral-related reads. Possible contaminating sequences or sequences that are not relevant can also be removed by aligning against reference sequences using several short reads with alignment tools such as BWA, SOAP2, and Bowtie2 (Langmead et al. 2012) (Fig. 2).
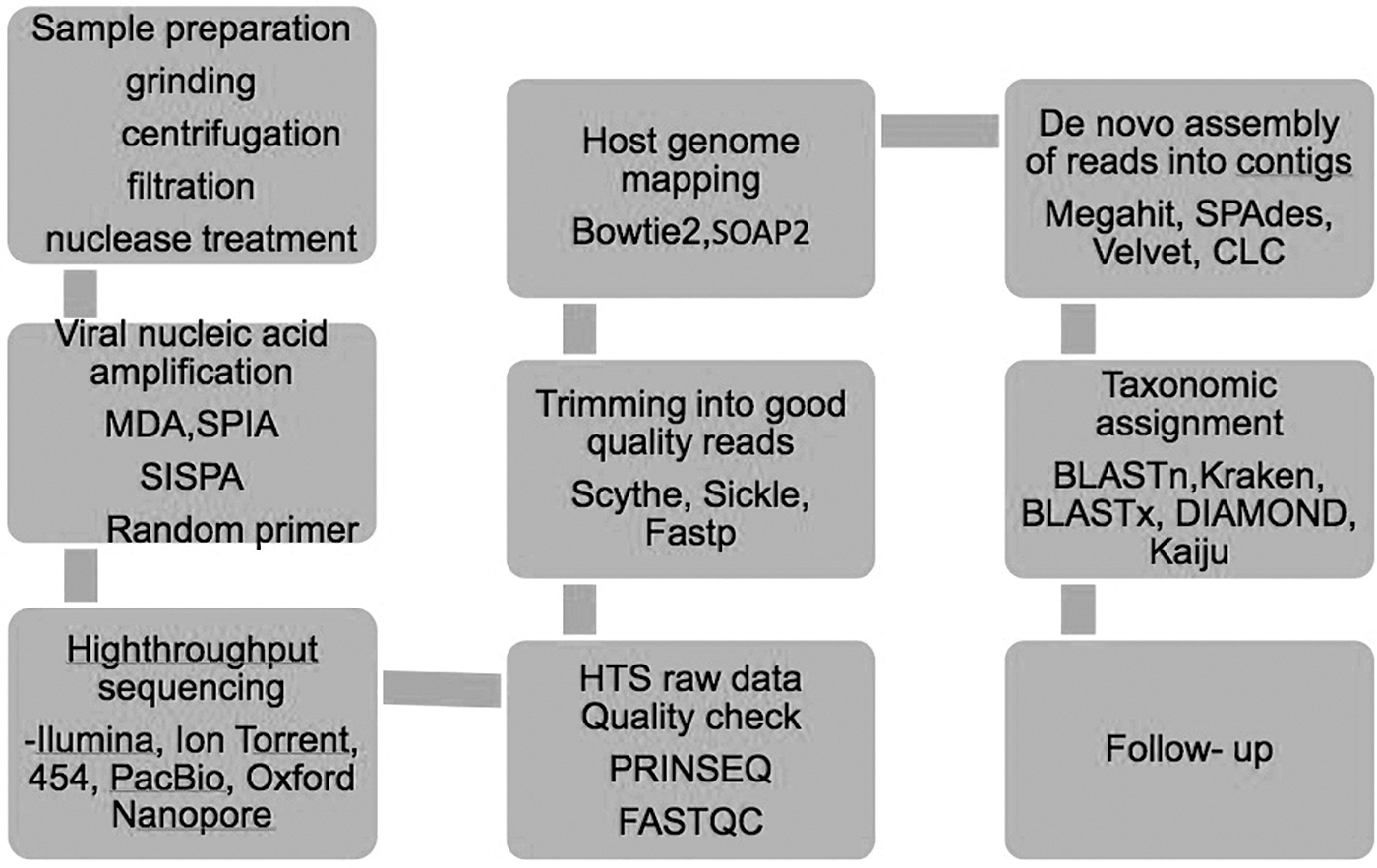
Detailed flow chart of procedures for metagenomic analysis of nucleic acids in tick samples.
De novo assembly
The assembly of shorter sequences that have matching overlaps generates longer sequences called contiguous sequences (contigs), a method referred to as de novo assembly. These contigs can be further extended by merging shorter contigs. There are two primary types of de novo assembly programs, Overlap/Layout/Consensus assemblers, including MIRA, Celera, and VICUNA, which are widely used for longer reads (Yang et al. 2012), and de Bruijn graph assemblers, such as Velvet, SOAP denovo, SPAdes, and MEGAHIT, (Fig. 2) which are widely used for shorter reads (Zerbino 2010; Bankevich et al. 2012). However, the assembly process can generate chimeric sequences involving the assembly of sequences from different organisms or species, which may be problematic in viral metagenomic studies as the biological sample may contain closely related viral sequences (Bankevich et al. 2012). Building longer contigs by assembling shorter reads can also sometimes be problematic because of the high viral diversity in the sample (Yang et al. 2012). Closely related viral genomes can be mapped to reference genome through reference-based assembly, which is computationally efficient; however, divergent viral genomes cannot be aligned by this approach (Bankevich et al. 2012). De novo assembly, used for reconstruction of full-length genomes, may generate ambiguous sequences due to mutations and recombination of closely related viruses (Yang et al. 2012). The de novo assembly process also generates complex assembly graphs and fragmented assemblies, which are computationally demanding.
Taxonomic classification
Taxonomic classification is the final step in the metagenomic analysis, wherein each sequence is assigned to a taxonomic group. Sequence classification is one of the most important issues in metagenomic studies and mainly depends on the similarity between the query sequence and annotated genomes in the database (Buchfink et al. 2015). The most commonly used similarity-based classification is the Basic Local Alignment Search Tool (BLAST) (Altschul et al. 1990), (Fig. 2) in which sequences are compared to known genomes. Different versions of BLAST can be used, such as BLASTx and tBLASTx. Considering the time span for sequence classification, several different tools such as RAPsearch2, DIAMOND, Kaiju, and Kraken have been developed, which can reduce the time required for analysis from weeks to days (Menzel et al. 2016). Classification programs based on nucleotide alignments and Kmer-based approaches such as BLASTn and Kraken, respectively, can have insufficient sensitivity for the detection of divergent viral sequences (Menzel et al. 2016).
Likewise, protein searches such as BLASTx, DIAMOND, and Kaiju (Fig. 2) can be slower and require powerful computers or highly optimized tools (Buchfink et al. 2015; Menzel et al. 2016). Customized databases, such as those containing only viral genomes, can be used for similarity-based searches, although reliance on such databases could result in sequence misclassification (Buchfink et al. 2015). Limited representation of viral sequences in sequence databases is another challenge for the classification of novel viruses since reads originating from viruses may be unclassified if the database does not include relevant viral sequences (Wood et al. 2014).
Follow-up studies
Viral metagenomics provide basic information about which viruses are present in a sample. However, more extensive analysis or follow-up studies are necessary to understand the roles of the identified virus/viruses (Blomstrom 2011). These analyses may include obtaining full-length viral genomes by the primer walking approach, Rapid Amplification of cDNA Ends analysis, virus isolation, and viral characterization and developing diagnostic assays, which all depend on the study objective (Blomstrom 2011).
Conclusion
Increasing anthropogenic activities have largely contributed to the current trends of EIDs. Among the effects of such activities is biodiversity loss, which is intrinsically intertwined with climate change. Both biodiversity loss and climate change have affected tick population structures and habitat boundaries in recent years. Because of changing ecological conditions and human encroachment into wildlife areas, new, as yet unidentified tick-borne viruses may emerge or strains of well-known tick-borne viruses could emerge, which have altered pathogenicity. Tick-borne viruses will continue to be prevalent and cause disease in humans and animals and will also continue to emerge and reemerge if conditions are suitable. Moreover, recent studies have continued to highlight the increased possibility of host-switching in ticks based on observations of zoonotic tick-borne pathogens in accidental hosts, apart from their normal reservoir hosts. Detection of the previously unknown Alkhuma virus in Saudi Arabia only a few years ago demonstrated such a host-switching event.
Although ticks are a major carrier and reservoir for novel viruses and have the potential to cause disease in wild and domestic animals, as well as humans across the world, information on viral diversity in tick species is limited. Active searches for new tick-borne viruses and tick-borne viruses of other families and genera are needed. Increased availability of data for different tick-borne viruses and various subtypes and strains may lead to a better understanding of the pathogenicity of these viruses. Due to difficulties in cultivation for many viruses and a lack of common viral gene markers, detection of viruses, particularly novel or uncharacterized viruses, using conventional, immunological, and PCR methods is very difficult. Besides the intensive work on the molecular biology of vector-borne viruses, more effort should be devoted to research on the ecology and natural cycles of these viruses for a better understanding of their transmission as well as fluctuations in natural transmission. The metagenomic approaches described in this review will provide important tools to investigate tick-borne viral diversity in different environments.
Footnotes
Acknowledgments
We are grateful to the numerous contributions to this article made by our colleagues. In particular, we thank the Swedish Government and Swedish International Development Cooperation Agency (SIDA) for contributions and financial support. We also extend special thanks to the University of Dar es Salaam, Tanzania, and Swedish University of Agricultural Sciences, Sweden, for supporting this article, as well as our colleagues and researchers at the Swedish University of Agricultural Sciences Global Bioinformatics Centre who provided valuable knowledge and skills concerning Bioinformatics and Metagenomics.
Author Disclosure statement
No conflicting financial interests exist.
Funding Information
No funding was received for this article.