
Abstract
The detection of novel or re-emergent pathogens necessitates the development of rapid, easy-to-use diagnostic tests that can be readily adapted and utilized in both clinical laboratories and field settings. Heartland virus (HRTV) is the first pathogenic Phlebovirus responsible for serious and fatal cases in the United States. We developed a qualitative test based on recombinase-polymerase-amplification coupled with lateral flow reading (RPA-LF) for rapid detection of HRTV. The RPA-LF detected HRTV with a limit of detection of 1.19–1.54 plaque-forming unit equivalents/reaction. In addition, the RPA-LF was able to detect 0.6075 copies/μL of HRTV nucleoprotein gene-containing plasmid. We evaluated six clinical samples that were previously found to be real-time PCR positive for HRTV and found five out of six samples to be positive by RPA-LF, yielding 83.3% concordance with real-time PCR. All six samples had Ct values between 29 and 39 by real-time PCR. We also determined that the HRTV primers and probe do not cross-react with other tick-transmitted viruses such as Bourbon and Powassan, or other related viruses, including Lonestar tick virus and Sunday canyon virus (100% specificity). This is the first isothermal amplification test developed for a tick-borne virus, which will allow for rapid differentiation between HRTV and other pathogens producing similar clinical manifestations.
Introduction
Heartland virus (HRTV) is a novel Phlebovirus in the family Phenuiviridae, which was first recognized in blood specimens obtained in 2009 from two very sick individuals residing at northwestern Missouri in the United States (McMullan et al. 2012). The patients had history of tick bite and were admitted to the hospital with nonspecific febrile illness with symptoms that included fever, fatigue, anorexia, and diarrhea. They also had common laboratory findings of leukopenia with moderate neutropenia, thrombocytopenia, and elevated hepatic aminotransferase levels. Both patients exhibited viremia on day 2 of hospitalization, ∼7 days after the onset of symptoms, and were empirically treated for ehrlichiosis, due to history of tick bite and the overlapping symptoms of leukopenia and thrombocytopenia (McMullan et al. 2012). Blood samples were sent to the Centers for Disease Control and Prevention (CDC) where ehrlichiae, rickettsiae, and borrelia infection were ruled out by culture, PCR, and serology (McMullan et al. 2012). At CDC, culture from both patients' leukocytes resulted in the isolation of HRTV. By using next-generation sequencing, it was determined that HRTV was a novel phlebovirus closely related to Huaiyangshan banyangvirus, also known as severe fever with thrombocytopenia syndrome virus (SFTSV), a potentially lethal tick-borne virus that causes hemorrhagic fever in Asia.
Since the isolation of HRTV, the CDC reported more than 40 cases of HRTV from the Midwestern and southern United States, including Tennessee, Missouri, Oklahoma, and Illinois, with three reported deaths (Muehlenbachs et al. 2014, Brault et al. 2018, Center for Disease Control and Prevention 2018a, Center for Disease Control and Prevention 2018b). The distribution of the HRTV cases mirrors closely the distribution of the lone star tick, Amblyomma americanum, and most patients reported finding a tick attached or crawling on their body before illness onset (Pastula et al. 2014, Brault et al. 2018). Laboratory transmission studies have confirmed the capacity of the lone star tick to transmit HRTV (Godsey et al. 2016). However, the reservoir hosts remain to be identified (Brault et al. 2018).
Seroprevalence studies of HRTV in blood donors of Northwestern Missouri estimated a seroprevalence of 0.9%, suggesting that several human infections have gone unidentified in this region (Lindsey et al. 2019). The results also highlight the need to test for HRTV patients with acute febrile illness and leukopenia and thrombocytopenia, who are suspected to have tick-borne disease and who do not improve after appropriate treatment (i.e., doxycycline). Given the largely nonspecific symptoms observed, HRTV could be a more common cause of human illness than is currently recognized.
Tick-transmitted diseases are on the rise in the United States, due to the expanding geographic range of the vectors of disease-causing pathogens (Saldarriaga et al. 2016). Different species of ticks have been implicated in the transmission of protozoa, bacteria, and viruses (Castellanos-Gonzalez et al. 2015, Saldarriaga et al. 2016). As many tick-borne illnesses have overlapping symptomology, development of rapid, point-of-care (POC) diagnostic assays is imperative. Simple, low-cost assays are beneficial for local laboratories, allowing them to perform initial differential diagnosis without having to send samples to larger regional or national laboratories. Molecular tests based on isothermal recombinase-polymerase-amplification coupled with lateral flow reading (RPA-LF) allow for detection of pathogen DNA or cDNA with similar sensitivities as qualitative real-time PCR, but without the need of expensive equipment (Castellanos-Gonzalez et al. 2015, Saldarriaga et al. 2016).
In this assay, to enable detection by lateral flow, the reverse primer is biotinylated at the 5′ end and there is an internal fluorescein amidite (FAM)-labeled probe included. The positive test band is produced when anti-biotin antibodies immobilize the amplified DNA, which contains the biotinylated reverse primers. The gold particles in the strip, which are covered with mouse anti-FAM antibodies, bind to the FAM-labeled probe making the test band visible. The reaction is validated by the appearance of the control band in the upper part of the strip. This band appears upon the immobilization of excess free-gold particles (which are covered with mouse antibodies) by means of anti-mouse antibodies. To date, the RPA-LF assays for cutaneous and visceral leishmaniasis (Castellanos-Gonzalez et al. 2015, Saldarriaga et al. 2016), intestinal protozoa (Cryptosporidium and Giardia) (Crannell et al. 2016), Entamoeba histolytica (Nair et al. 2015), and Trypanosoma cruzi (Jimenez-Coello et al. 2018) have shown similar sensitivity and specificity as the respective real-time or conventional PCR assays. In this study, we report the development of the RPA-LF test for the rapid detection of HRTV from clinical samples and evaluate its efficacy in comparison with real-time PCR testing. This molecular assay could be implemented in decentralized laboratories to differentiate HRTV from other tick-borne pathogens with similar symptomologies that could be geographically overlapping (McMullan et al. 2012).
Materials and Methods
Viruses
Viruses used in this study were originally obtained from the World Reference Center for Emerging Viruses and Arboviruses at University of Texas Medical Branch (UTMB) and expanded in the investigators' laboratories to generate virus stocks, obtain virus titers, and generate viral RNAs for the RPA-LF reactions.
Patient samples
Blood samples obtained from patients with acute undifferentiated febrile illness were initially screened by RT-PCR for HRTV at the Tennessee Department of Health. De-identified samples that were suspected to contain HRTV genomic material were sent to UTMB for further laboratory analysis under UTMB Institutional Review Board protocol #95-111.
Viral RNA isolation and cDNA synthesis
HRTV was titrated on Vero cells by plaque assay using previously described methods (Juarez et al. 2013). Virus stocks were spiked in serum and diluted 10-fold to evaluate assay sensitivity. Virus was inactivated by using Trizol followed by modified chloroform separation and RNA isolation using the Qiagen RNeasy Mini Kit (Qiagen). The cDNA synthesis was carried out using the iScript Select cDNA Synthesis Kit (Bio-Rad) following the manufacturer's protocol. The cDNA synthesis reactions included 10 μL of RNA and either 3 μL of random primers (Bio-Rad) or HRTV-specific primers (5 μm) targeting the nucleoprotein (NP) gene. The primers used in the cDNA synthesis are detailed in Table 1. Virus-specific primers were evaluated individually, in pairs, or as a mix of four and six primers in the cDNA synthesis reaction to identify the condition that was most effective in enhancing the sensitivity of the RPA-LF reaction.
Polymerase Chain Reaction and Recombinase-Polymerase-Amplification Primers and Probe
RPA-LF, recombinase-polymerase-amplification coupled with lateral flow reading.
Quantitative real-time PCR for HRTV
The real-time quantitative PCR (qPCR) assay for HRTV was used to assess and confirm viral presence in clinical samples and was carried out using previously described methods (Savage et al. 2013). Briefly, the PCR was performed using the iScript One-Step Kit (BioRad, CA) and the primers HRTV1-Forward TGCAGGCTGCTCATTTATTC, HRTV1-Reverse CCTGTGGAAGAAACCTCTCC, and HRTV1-probe FAM-CCTGACCTGTCTCGACTGCCCA-TAMRA. Reactions of 20 μL were carried out using a StepOne Plus system (Applied Biosystems). Cycling was as follows: 1 cycle of 50°C for 10 min followed by 95°C for 1 min and 40 cycles of 95°C for 15 s followed by 60°C for 30 s.
HRTV primer design and position using Visual OMP software
Since the primers available in the literature (Savage et al. 2013) and used in the quantitative RT-PCR showed poor amplification efficiency under isothermal conditions (data not shown), we designed primers using the vOMP software (DNA Software, Ann Arbor, MI), which provides rapid in silico assessment of primer binding efficiency to target sequence. The program also predicts the formation of primer dimers and homodimers, indicating at what temperatures secondary structures may occur under specific reaction conditions. To enable for detection by lateral flow, the reverse primer was biotinylated at the 5′ end. We designed a 45 bp conserved internal probe (IDT, Petaluma, CA) that included FAM (5′-carboxy fluorescein amidite) at the 5′ end, an internal dSpacer, and a SpacerC3 in the 3′ end, as suggested by the manufacturer (TwistDx, United Kingdom). One of two primer pairs that we designed targeting the nucleoscapsid (NP) gene of HRTV consistently amplified the virus, showing clear positive bands in the lateral flow strips. Table 1 shows the primers and probe sequences used in the RPA-LF assay.
RPA reaction and LF reading
The amplification mixture comprised the following: (1) forward primer (2.4 μL, 5 μM), (2) biotinylated reverse primer (2.4 μL, 5 μM), (3) FAM-labeled probe (0.3 μL, 5 μM), (4) magnesium acetate (1.25 μL, 288 mM), (5) Betaine (3.3 μL, 3 M stock in water), and (6) the rehydrated cocktail (Twist amp nfo RPA kit; TwistDx). Viral cDNA (2.5 μL) was immediately added to the mixture and subjected to amplification at 43.5°C for 40 min using a dry bath incubator (VWR). The RPA product was diluted 1:50 in 100 μL of dipstick assay buffer in a 1.5 μL Eppendorf tube. The bottom tip of the lateral flow strip (Ustar Biotechnologies, Hangzhou) was then immersed in the sample, making the amplification product run upwards by capillarity. Viral cDNA amplification was confirmed with the naked eye after 5 min by the appearance of the test band in the lower part of the strip.
Statistical analysis
Probit analysis was conducted for the lowest concentrations of detected analytical virus samples. Statistical analysis was done using free software.
Results
Assay design
The nucleocapsid gene (NP) is one of most conserved viral proteins of HRTV, and therefore, primers were designed targeting this conserved viral gene (Brault et al. 2018). Using the primers designed by the vOMP software (DNA Software, Ann Arbor, MI), we did a blast search (National Center for Biotechnology Information) to ensure that the primers designed by the program would target known NP sequences of HRTV and would not cross-react with other viruses and no other organisms, including humans and ticks. Only HRTV sequences returned in our searches.
HRTV RPA-LF specificity
The specificity of the HRTV RPA-LF was tested against a panel of viral RNA extracts, encompassing both phylogenetically related viruses (SFTSV), viruses transmitted by the same vector A. americanum (Bourbon bayou virus [BRBV] and Lone star tick virus [LSV]) and viruses with overlapping geographic distribution (Powassan virus [POWV] and Sunday canyon virus [SCAV]). We found no cross-reactivity of the HRTV RPA-LF test with any other virus RNA extract supporting the specificity of the HRTV RPA-LF assay (Fig. 1). Each RPA-LF was exposed to ∼500 plaque-forming unit (pfu) equivalents/reaction or more, of each virus respectively, and as expected, only HRTV was amplified (n = 9 [HRTV] and 5 [BRBV, LSV, POWV, SCAV, and SFTSV], XLSTAT contingency analysis p = 0.0003).

RPA-LF specificity. Relevant viruses were tested for their cross-reactivity within our diagnostic assay. All viral samples were prepared like the HRTV controls. HRTV, Heartland virus; POWV, Powassan virus; BRBV, Bourbon bayou virus; LSV, Lone star tick virus, SCAV, Sunday canyon virus; SFTSV, severe fever with thrombocytopenia syndrome virus; RPA-LF, recombinase-polymerase-amplification coupled with lateral flow reading.
Analytical sensitivity
In initial optimization, we compared the efficiency of viral RNA to cDNA synthesis using virus-specific primers or random primers. For the cDNA synthesis, we evaluated the efficacy of the reaction using a single primer, primer pairs, or a mix of four and six primers. We found that while all combinations of primers were relatively effective at converting viral RNA (data not shown), the mix of four reverse primers resulted in enhanced sensitivity of the RPA-LF assay (Fig. 2A).

Analytical efficacy of HRTV RPA-LF. Efficiency of RNA to cDNA conversion using gene-specific reverse primer (+) or random primers (
To test the sensitivity of the RPA-LF assay, we serially diluted the viral RNA and carried out the cDNA synthesis, and the resulting cDNA was used in the RPA-LF assay in 3–5 replicates. Although both random primers and virus-specific primers generated cDNA that was successfully detected in the RPA-LF reaction, the virus-specific primers enhanced the sensitivity of the RPA-LF assay by two-fold and allowed us to detect 1.19 pfu equivalents/reaction versus 2.38/reaction when using the random primers (Fig. 2A).
To further evaluate the sensitivity, the assay was tested with HRTV-spiked serum samples to determine whether serum components could negatively impact the amplification efficiency of RPA-LF. For this purpose, we serially diluted known concentrations (pfu) of HRTV in serum, extracted RNA, and carried out cDNA synthesis using virus-specific primers and RPA-LF assay. The results demonstrated a similar limit of detection to that obtained with serial dilutions of virus suspended in culture medium. The limit of detection in serum samples was 1.54 pfu equivalents/100 μL or 15.4 pfu/mL of blood (n = 4) and all negative samples were negative (n = 6). Probit analysis generated a p-value of 0.0009, indicating that the possibility of false positives is null (Fig. 2B).
In addition to serially diluted virus in serum, we assessed the sensitivity of the HRTV RPA-LF using a plasmid containing the NP gene, allowing the direct quantification of copies/μL/reaction. Known concentrations of plasmid DNA were serially diluted in water until we were no longer able to detect signal by RPA-LF. Using known concentrations of plasmid, we performed multiple RPA-LFs (n = 5) compared to cDNA from uninfected serum as a negative control to determine the limit of detection in copies/μL (Fig. 3). We were able to detect between 0.972 and 0.0972 copied/μL; band intensity was diminished, but faintly observable at these concentrations (Fig. 3, Red box). Additional dilutions were performed, and we were able to repeatedly detect diluted plasmid to as low as 0.6075 copies/μL (n = 5, p = 0.000197, indicating the likelihood of false positives is null).

LOD NP plasmid. HRTV NSP plasmid was serially diluted and RPA-LF performed as described to determine the LOD. Strong bands were observed through 9.72 copies/μL. Fainter bands were detected at 0.972 and 0.0972 copies/μL (Red box) with no bands appearing in the nonspecific DNA controls. Additional concentrations between 9.72 and 0.972 copies/μL were tested to determine the repeatability of the assay (n = 5).
Diagnostic efficacy of HTRV RPA-LF
Given the results described above, we proceeded to test the assay using six clinical samples previously found to be HRTV positive by real-time PCR. We extracted RNA from 100 μL of serum and converted to cDNA using a gene-specific primer or random primers. The RPA-LF found five out of the six samples to be HRTV-positive yielding, while all samples were positive by real-time PCR. All positive tests showed lighter bands in the lateral flow strips compared with the positive control (Fig. 4), consistent with the relatively low viral loads present in the samples as determined by the high Ct values (≥29) (Table 2).
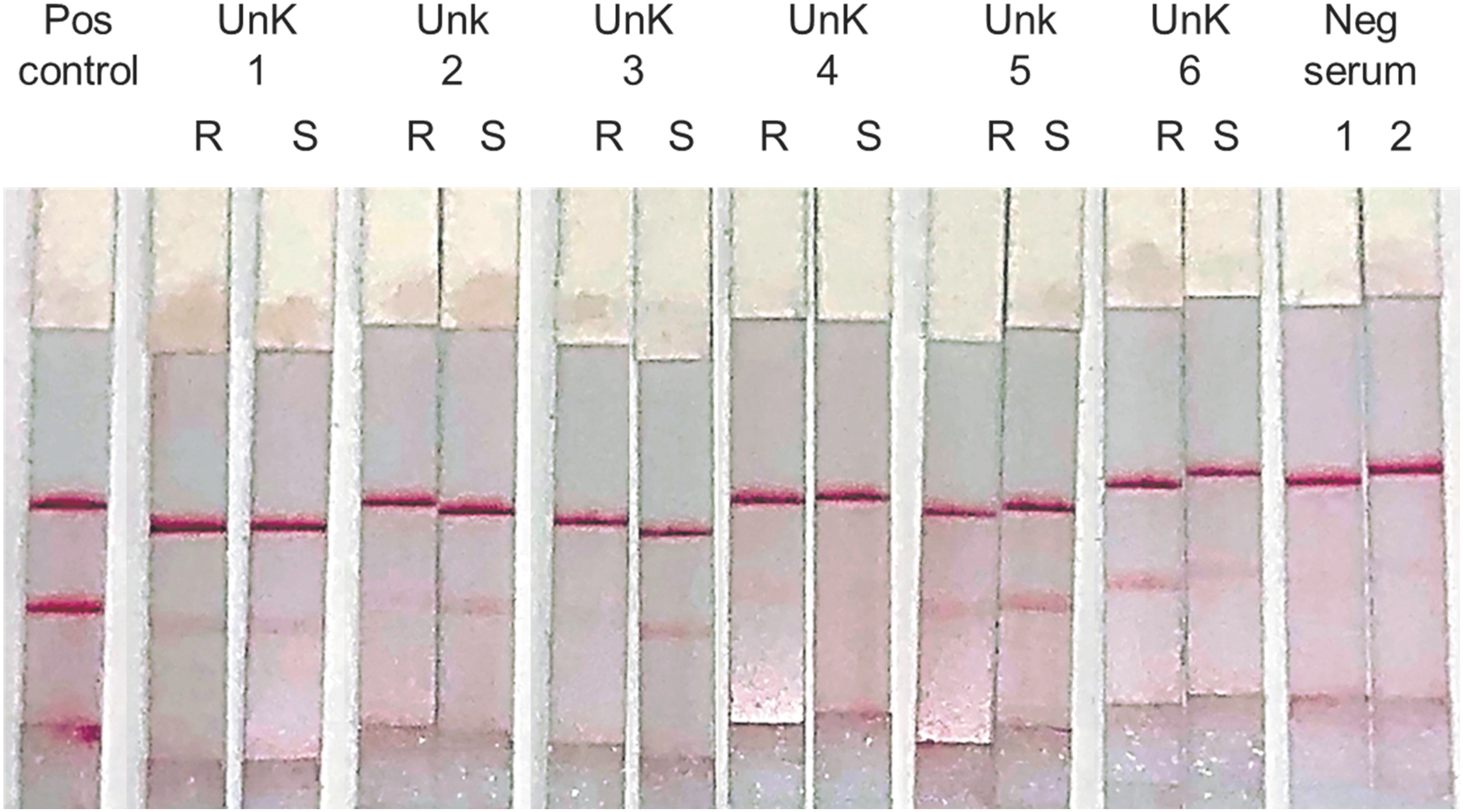
Clinical efficacy of HRTV RPA-LF. Assessment of diagnostic efficacy using gene-specific primer (S) or random primers (R) to detect HTRV cDNA in human serum samples. Light positive bands were detected in all samples, except in #4. Utilization of gene-specific primer for RNA to cDNA synthesis produced clearer bands, except in sample #6.
Comparison of Recombinase-Polymerase-Amplification Coupled With Lateral Flow Reading and RT-Quantitative Polymerase Chain Reaction Results
+++: strong reaction; ++: evident band; +: faint-light band; −: negative reaction, no band.
N.D., not determined.
Discussion
The expansion of tick ranges and emergence of new infectious agents make development of accessible and inexpensive diagnostic assays paramount to the diagnosis and treatment of transmissible diseases (McMullan et al. 2012). Currently, there is no rapid test for HRTV, and most clinical laboratories do not have the capabilities to perform a qualitative or qPCR for this virus.
The availability of a POC molecular test such as RPA-LF could contribute to the diagnosis of febrile undifferentiated illness and increase the epidemiological information of this tick-borne pathogen. The development of a POC assay that can rule out HRTV infection after tick exposure is an important step in tick-transmitted control. Furthermore, its implementation in laboratories from regions suspected to be endemic for this virus could help define the geographical distribution and infection rates of humans and vectors of HRTV.
In this study, we describe the development and assessment of a rapid POC diagnostic test for the tick-borne HRTV. We were able to detect low concentrations of HRTV using virus-spiked serum, resulting in strong signal in the LF strips. However, the band intensity of the positive clinical samples was much lighter compared with the lateral flow strips, even with those corresponding to 1.54 pfu/100 μL. The assumption that patient samples harbored low HRTV RNA was later confirmed by the high Ct values of the qPCR (Table 2). The clinical samples used in this study were not evaluated immediately after their collection and probably have been frozen and thawed several times before our handling. Typically, clinical samples, when tests are available, are processed immediately, without any freezing or long-term storage issue that may affect test results. Consequently, it is likely that a significant proportion of RNA was already degraded. This potential degradation could be caused by physical shearing of the RNA due to repetitive ice crystal formation, oxidation of the nucleic acids, or the presence of degradative enzymes (Roche Life Science 2018). This could result in the cleaving of viral RNA, thus reducing the potential binding sites for our larger RPA primers, while leaving the RT-qPCR's smaller targets intact, potentially explaining the disagreement between the RPA-LF and RT-qPCR result for Unknown #4 (Fig. 4; Table 2) (Kopreski et al. 1999, Oliveira et al. 2012). Nevertheless, despite the aforementioned drawback, our RPA-LF was able to detect 5/6 previously confirmed HRTV infections.
The primers and probe designed for the HRTV RPA-LF have a 100% specificity. When tested against genetically related viruses or those that are transmitted by the same vector, there was no evidence of cross-reactivity.
Conclusions
This assay will provide a unique tool for rapid screening of suspected tick-borne pathogens in endemic regions and allow for proper patient treatment without having to send clinical samples to the CDC or other public health laboratories for initial diagnosis.
Footnotes
Acknowledgments
We would like to thank the World Reference Center for Emerging Viruses and Arboviruses at University of Texas Medical Branch for providing viral samples to test in our assay and the Western Gulf Coast Center of Excellence for support.
Authors' Contributions
Conception or design of the work: T.R.S., P.V.A., and B.L.T. Data collection: T.R.S. and N.E.B. Provision of viral stock and reagents: E.S.R. and S.T. Provision of clinical samples: A.M. and L.B. Data analysis and interpretation: T.R.S., N.E.B., P.V.A., and B.L.T. Drafting the article: T.R.S., N.E.B., P.V.A., and B.L.T. Critical revision of the article: T.R.S., A.M., K.B., S.T., P.V.A., and B.L.T. Final approval of the version to be published: T.R.S., N.E.B., A.M., E.S.R., S.T., P.C.M., K.B., P.V.A., and B.L.T.
Author Disclosure Statement
No conflicting financial interests exist.
Funding Information
This work was supported through cooperative agreement number 1U01CK000512 between the Centers for Disease Control and Prevention (CDC) and University of Texas Medical Branch/Western Gulf Center for Excellence in Vector-Borne Diseases.