
Abstract
Rickettsiae and bartonellae are Gram-negative bacteria that can cause zoonotic and human diseases and are vectored by hematophagous arthropods. In the Americas, rickettsioses and bartonelloses have reemerged as significant public health threats. Bartonella species have been identified as causing zoonotic infections responsible for a variety of clinical syndromes in humans and animals. The aim of this study was to investigate the distribution, prevalence, and molecular heterogeneity of Rickettsia spp. and Bartonella spp. among ectoparasites collected from domestic animals in 14 farming communities in the Andes Mountains of Cuzco, Peru. A total of 222 domestic animals representing 8 different species (sheep, donkeys, goats, cattle, pigs, llamas, guinea pigs, and horses) were sampled. Nine species of ectoparasites (n = 1,697) collected from 122 animals were identified resulting in 1,657 chewing lice, 39 ticks, and 1 flea. DNA was individually extracted from a random sample of 600 (35.4%) considering variability of ectoparasite species, hosts, and sample location elevation. All 600 samples were negative for rickettsial DNA by a genus-specific molecular assay. A subset of 173 (28.8%) samples were selected based on variability of arthropods species, host, and location for Bartonella testing. Ninety-one (52.6%) of these samples including Melophagus ovinus (90/110) and Bovicola bovis (1/7) were positive for Bartonella by a genus-specific molecular assay. Five Bartonella genes of seven DNA samples from M. ovinus were analyzed by the multilocus sequence typing for characterization. We identified five identical Bartonella melophagi specimens and two specimens with Bartonella species related to B. melophagi from the seven M. ovinus. The Bartonella agents detected were widely distributed and frequent in multiple studied locations.
Introduction
Rickettsiae and bartonellae are Gram-negative bacteria that can cause zoonotic diseases and are vectored by hematophagous arthropods such as mites, ticks, sandflies, kissing bugs, biting/chewing and sucking lice ectoparasites (Angelakis et al. 2010, Chomel and Kasten 2010, Mullins et al. 2013, Kumsa et al. 2014, Kosoy et al. 2018, Yoshimizu and Billeter 2018).
Bartonella species have been identified as important zoonotic pathogens transmitted by variety of arthropods (Parola et al. 2002, Billeter et al. 2011, Regier et al. 2016, Laroche et al. 2017, Do Amaral et al. 2018) and are responsible for an assortment of clinical presentations in humans, and may cause paucisymptomatic bacteremia and endocarditis (Maggi et al. 2009, Regier et al. 2016, Vayssier-Taussat et al. 2016). Approximately 15 species of Bartonella have been isolated or sequenced from human samples (Diniz et al. 2013). Domestic and wild animal are potential reservoirs of Bartonella and Rickettsia species (Chang et al. 1994, Chomel et al. 2006, Kumsa et al. 2014, Gutierrez et al. 2015, Kho et al. 2015, Promrangsee et al. 2019, Salmon-Mulanovich et al. 2019) where human health and animal health are closely linked.
Approximately 75% of emerging infectious diseases are zoonotic and 28% are vector-borne (Regier et al. 2016). Thus, the need for a one-health approach to reduce the risk of zoonotic diseases among humans and animals (Regier et al. 2016). Vector-borne zoonotic diseases are also a threat to military operations around the world, among which are rickettsioses (e.g., Rocky Mountain spotted fever, Tidewater spotted fever, epidemic typhus, flea-borne spotted fever) and bartonellosis (e.g., trench fever, Bartonella spp.) (Chomel et al. 2012, Garcia et al. 2017, Davis et al. 2020). In studies from the Americas, various genera of hematophagous ectoparasites from domestic and wild animals were found infected with Rickettsia spp. and Bartonella spp. (Parola et al. 2002, Wikswo et al. 2007, Billeter et al. 2011, Caceres et al. 2013, Fontalvo et al. 2017, Do Amaral et al. 2018, Rizzo et al. 2019).
This study was designed to determine the distribution, prevalence, and molecular heterogeneity of Rickettsia spp. and Bartonella spp. in ectoparasites that were collected from domestic animals in 14 farming communities in Cuzco, Peru.
Materials and Methods
Collection sites
Sampling was conducted at 14 sites in the Department of Cuzco (Table 1), which is in the high Andean region of southern Peru. The altitude of the collection sites was between 2,938 and 4,014 meters above sea level. Field work was performed during the winter season in 1 week of August 2013. At the sample collection time, the average minimum and maximum temperatures were 2°C (at night) and 24°C (at midday), respectively.
Locations of Domestic Animals Assessed for Ectoparasites in the Department of Cuzco, Peru
MASL, meters above sea level.
Ectoparasite collection
A total of 222 individual subjects were examined representing 8 different species of domestic animals: sheep (117), donkeys (16), goats (3), cattle (71), pigs (7), llamas (1), guinea pigs (2), and horses (5) (Table 1). All visible ectoparasites were removed with fine forceps and placed in labeled cryovials (1.8 mL) containing 95% ethanol and transported to the Naval Medical Research Unit No. 6 in Lima, Peru. Ectoparasites were identified using morphological keys (Price and Graham 1997, Goddard and Layton 2006).
All collections were conducted under the auspices of Resolución Directoral No. 0406-2013-MINAGRI-DGFFS/DGEFFS; molecular screening in ectoparasites was conducted under the auspices of Contrato de Acceso Marco a Recursos Genéticos No. 0017-2014 MINAGRI-DGFFS/DGEFFS.
Ectoparasite testing
Ectoparasites previously washed with sterile water were cut in half using sterile scalpels. Genomic DNA (gDNA) was extracted individually from one-half of each ectoparasite by mechanical disruption using a Kontes Pellet Pestle™ (Fisherbrand™) in 100 μL of PrepMan™ Ultra Sample Preparation Reagent (Applied Biosystems™). The other half of the arthropod was stored at −30°C as a backup. Crushed individual ectoparasite samples were heated to 95°C for 10 min using a heat block and centrifuged at room temperature for 5 min at top speed (13,000 rpm) using a tabletop Eppendorf® centrifuge (Eppendorf AG). Clear supernatants were transferred to clean tubes and stored at −20°C until further processing.
Ectoparasite DNA samples (n = 600) were selected based upon variability of arthropod species, host and sample locations for the detection of Rickettsia species. Aliquots of five individually extracted DNA samples were pooled together as previously described (Kocher et al. 2016) for the screening of Rickettsia spp. using a quantitative real-time PCR (qPCR) assay (Rick17b) that targets the conserved 17 kDa Rickettsia antigen gene (Jiang et al. 2012).
Subsequently, a subset of 173 (28.8%) samples were randomly selected from the previous 600 ectoparasite DNA samples using the same sample criteria for testing Bartonella. Individual DNA samples were tested by the genus-specific qPCR assay (BartAG) with primers and specific probe to detect the presence of Bartonella ftsZ (forward primer 5′GTGYTTTATTCTGGTGTTGCTTC3′, reverse primer 5′GCAATAGCAGCTTCAGCMG3′ and the probe 5′-FAM-TGCWGATGTTCGYTCTGTTATGCATGAAATGG-BHQ-1-3′).
The BartAG qPCR reactions were run using Supermix-UDG and 6 mM MgCl2. Each reaction contained 0.7 μM of the forward and reverse primers and 0.4 μM of the probe. In addition, 3 μL of template DNA was added to each reaction. The cycler parameters included an initial incubation at 50°C for 2 min followed by an initial denaturation at 95°C for 2 min, then 45 cycles of denaturation at 95°C for 15 s and annealing/elongation at 60°C for 30 min using a StepOne™ Real-Time PCR System (Applied Biosystems).
After optimization of the BartAG qPCR assay, sensitivity (using 16 Bartonella species DNA) (Table 2) and specificity (using 17 non-Bartonella species DNA) (Table 3) were determined to be 100% each (Broeders et al. 2014). The limit of detection of the BartAG assay was determined by serially diluting each of the Bartonella species (Table 2) 10-fold with molecular grade water down to 1 copy/μL. The limit of detection was ascertained to be 1 copy/μL for each of the 16 Bartonella species assessed. Linear DNA (LBartGAn) was used as a positive control.
Sensitivity and Limit of Detection of the New Bartonella Genus-Specific Quantitative Real-Time PCR Assay: BartAG
Genome size estimated to be 2.0 Mb.
Ct, cycle threshold; qPCR, quantitative real-time PCR.
Specificity of the New Bartonella Genus-Specific Quantitative Real-Time PCR Assay: BartAG
We selected seven samples that were positive for Bartonella qPCR for characterization at the species level using the multilocus sequence typing (MLST) based on five genes, the citrate synthase (gltA), RNA polymerase B-subunit (rpoB), cell division protein (ftsZ), 16S ribosomal RNA (rrs), and the 60 kDa heat shock protein (groEL) genes. All genes were amplified using conventional PCR and nested PCR as previously described (Mullins et al. 2013). PCR amplifications were run on a thermocycler (ARTICK5020; Thermo Fisher Scientific, Ratastie, Finland). PCR products were separated by electrophoresis in 1.5% agarose gels and then purified using ExoSAP-IT™ (Applied Biosystems), and subsequently sequenced using Big Dye terminator kit in the 3130XL Genetic Analyzer (Applied Biosystems).
The MLST analysis was conducted against known Bartonella species available in the GenBank database and was based on the comparison with full or partial gltA, ftsZ, rrs, rpoB, and groEL genes. To optimize the MLST analysis, we used the BLAST highly similar option to optimize the detection of Bartonella including the name of the Bartonella species of interest. To infer phylogenetic relationships among available well-characterized Bartonella species and the seven Bartonella spp. described herein, we first aligned the sequences with MUSCLE and then built phylogenetic trees using concatenated genes with the maximum likelihood algorithm considering 2,000 bootstrap replicates in MEGA X Version 10.0.5 (Kumar et al. 2018).
The phylogenetic analysis, which included genetic distances, was calculated using the Hasegawa–Kishino–Yano model (Hasegawa et al. 1985), which was selected among other substitution models for the likelihood ratio test. Partial genes sequences generated in this study were submitted to GenBank under the following accession numbers: nonribosomal sequences, MT154626–MT154653 and 16s RNA ribosomal sequences, MT152264–MT152270.
To further the analysis of the Bartonella spp. detected, the partial genomes were sequenced by next-generation sequencing using nanopore technology from eight DNA samples with a standard whole-genome amplification approach. Nucleic acid concentrations were determined using an Agilent 4200 Tapestation (Agilent, Inc., Santa Clara, CA). Ten nanograms of each sample was amplified using Phi29 polymerase (Repli-g midi kit; Qiagen) overnight at 16°C and then DNA knots were removed by endonuclease H digestion (New England Biolabs, Ipswich, MA) for 15 min at 30°C and subsequent DNA purification using the ZymoResearch silica spin column gDNA kit (ZymoResearch). Following the manufacturer's protocol, individual samples were barcoded by ligation (ONT NBD-104), purified on AMPure XP beads, and then quantified on the Tapestation.
About 160 ng of four individual barcoded libraries were combined to produce a multiplexed sample that was prepared for sequencing using the LSK-109 kit (ONT) and then applied to primed R9.4 RevD flow cells on a GridION sequencer (ONT). Data were collected continuously during the 48-h run and base called using the high accuracy GUPPY basecaller function (v3.0.3) by MinKNOW. Resulting FASTQ data from each run were demultiplexed and compared with available Bartonella species using the online EPI2Me platform (
Results
From 122 animals (106/117 sheep, 1/16 donkeys, 2/3 goats, 8/71 cattle, 4/7 pigs and 1/5 horses), a total of 1,697 arthropods (1,657 chewing lice, 39 ticks, and 1 flea) were collected (Table 1). Two of the animals examined, guinea pigs (n = 2) and llamas (n = 1), did not yield any ectoparasites.
Among the arthropods collected various species of biting/chewing lice were included: Melophagus ovinus, the sheep ked or chewing louse (1,563); Bovicola bovis, red louse or cattle biting louse (12); Bovicola caprae, goat biting louse (4); Bovicola equinus, horse biting louse (10); Haematopinus eurysternus, short-nosed sucking louse of cattle (5); Haematopinus suis, hog louse (44); and Linognathus stenopsis, sucking goat louse (19). Other ectoparasites collected included: the soft tick Otobius megnini, spinose ear tick (39); and Tunga penetrans the jigger flea (1). Species of ectoparasites collected are given in Table 4.
Detection of Rickettsia spp. and Bartonella spp. in Ectoparasites Collected from Domestic Animals by Genus-Specific qPCR Assays
Geographical sampling collection according Table 1.
All Rick17b qPCR assay results were negative; positive controls for each assay were positive.
Cow infested with two species of ectoparasites.
Goats infested with two species of ectoparasites.
M. ovinus was the most common ectoparasite (92%, n = 1,563), and was found on infested sheep (91.3%, n = 1,551) (Table 5) and donkeys (0.7%, n = 12). The other 10 species of ectoparasites made up only 8% of all the ectoparasites collected on domestic animals (Table 4). M. ovinus was the most common ectoparasite gathered for all collection sites.
Detection of Bartonella spp. in Melophagus ovinus Collected from Sheep at 14 Sites in the Department of Cuzco
A total of 600 samples were selected for rickettsia testing using a genus-specific qPCR assay (Rick17b): B. bovis (8/12), B. caprae (3/4), B. equinus, (8/10), H. eurysternus (3/5), H. suis (22/44), L. stenopsis (15/19), M. ovinus (505/1,563), O. megnini (35/39), and T. penetrans (1/1). All these samples were negative for rickettsial DNA (Tables 4), although the positive control consistently provided positive results.
Approximately one-third (28.8%, 173/600) of samples that were negative for Rickettsia spp. were screened for Bartonella by a genus-specific qPCR assay (BartAG). The following ectoparasites species were included: B. bovis (7/8), B. caprae (2/3), B. equinus, (3/8), H. eurysternus (2/3), H. suis (6/22), L. stenopsis (7/15), M. ovinus (110/505), O. megnini (35/35), and T. penetrans (1/1) (Tables 4 and 5). Bartonella spp. DNA were identified in 91 samples with cycle threshold (Ct) values ranging from 14.7 to 29.8 (mean = 21.4).
The positive samples belonged to two species of ectoparasites, M. ovinus and B. bovis. The prevalence of Bartonella spp. in the 173 DNA samples was 52.6% (91/173). The prevalence of Bartonella spp. in M. ovinus was 81.8% (90/110) and 14.3% (1/7) in B. bovis. The Bartonella agents were detected in all collection sites (Table 4). A total of 91 ectoparasites from 23 domestic animals from 14 sites were positive for BartAG qPCR. In the Molle Molle site two different ectoparasite infestations among a single goat (B. caprae and L. stenopis) and a single cow (B. bovis and O. megnini) were observed. All those ectoparasites tested were negative for BartAG qPCR (Table 4).
To characterize Bartonella spp. by MLST, 7 of 91 Bartonella-positive samples were selected. This subset of M. ovinus samples was representative of five different farming communities (Table 6). All five MLST genes were amplified from the seven samples (R-20-9, R-21-1, R-30-8, R-43-5, R-120-8, R-122-4 and R-161-3). The average length calculated for each MLST genes utilized was 335 base pairs (bp) for gltA, 786 bp for ftsZ, 824 bp for groEL, 600 bp for rpoB, and 263 bp for rrs.
Multilocus Sequence Typing and Percent Identity of Bartonella Gene Fragments gltA, ftsZ, rrs, and rpoB Among Known Bartonella Species to Bartonella DNA Specimens from Cuzco, Peru
All Bartonella species characterized in this study had identical gltA, ftZ, groEL and rrs genes. Comparisons were made using consensus sequences of gltA (353 bp), ftsZ (708 bp), groEL (737 bp) and rrs (208 bp) against known Bartonella species available in GenBank.
rpoB gene fragments were identical among strains R-21-1, R-30-8, R-43-5, R-120-8 and R-161-3. Comparisons were made using consensus sequences of 583 bp against known Bartonella species in GenBank.
rpoB gene fragments of strains R-20-9 and R-122-4 were identical. Comparisons were made using consensus sequences of 598 bp against known Bartonella species in GenBank.
bp, Base pairs; N.A., not available; N.S., sequences available but did not have significant identity match due to short alignments (e > 0).
All Bartonella positive specimens characterized in this study had identical gltA, ftsZ, groEL, and rss gene sequences. The MLST analysis suggests that our specimens were highly similar to B. melopaghi strain K-2C and K-9B when the gltA, ftsZ, and rpoB genes were analyzed. We additionally identified a high identity percentage to the Bartonella sp. Pangjai-1 when the groEL gene was analyzed. We were unable to compare our strains with Bartonella melophagi as its groEL sequence is not available in GenBank. The observed power of discrimination among Bartonella species was not good owing to several hits with 100% of identity to the strains described in this study (Table 7).
Identification and Characterization of Bartonella Species from Melophagus ovinus Collected from Sheep, Cuzco, Peru
Phylogenetic analyses included all sequenced samples and was conducted based on the concatenated sequences of three (gltA, ftsZ, and rpoB resulting in 1,601 nucleotides) and four (gltA, ftsZ, rrs, and rpoB resulting in 1,809 nucleotides) genes. The phylogenetic tree with four concatenated sequences displayed better support to establish that R-20-9 and R-122-4 are B. melophagi variants in comparison with the tree with three concatenated sequences. In general, both phylogenetic analyses were consistent suggesting that all strains studied here are closely related to the B. melophagi K-2C. Specifically, five strains (R-21-1, R-30-8, R-43-5, R-120-8, and R-161-3) were identical to the prototype B. melophagi strain K-2C and the other two strains (R-20-9 and R-122-4) were less similar but also related to the same prototype (Fig. 1). We did not identify patterns of geographical distributions among the sequenced samples.
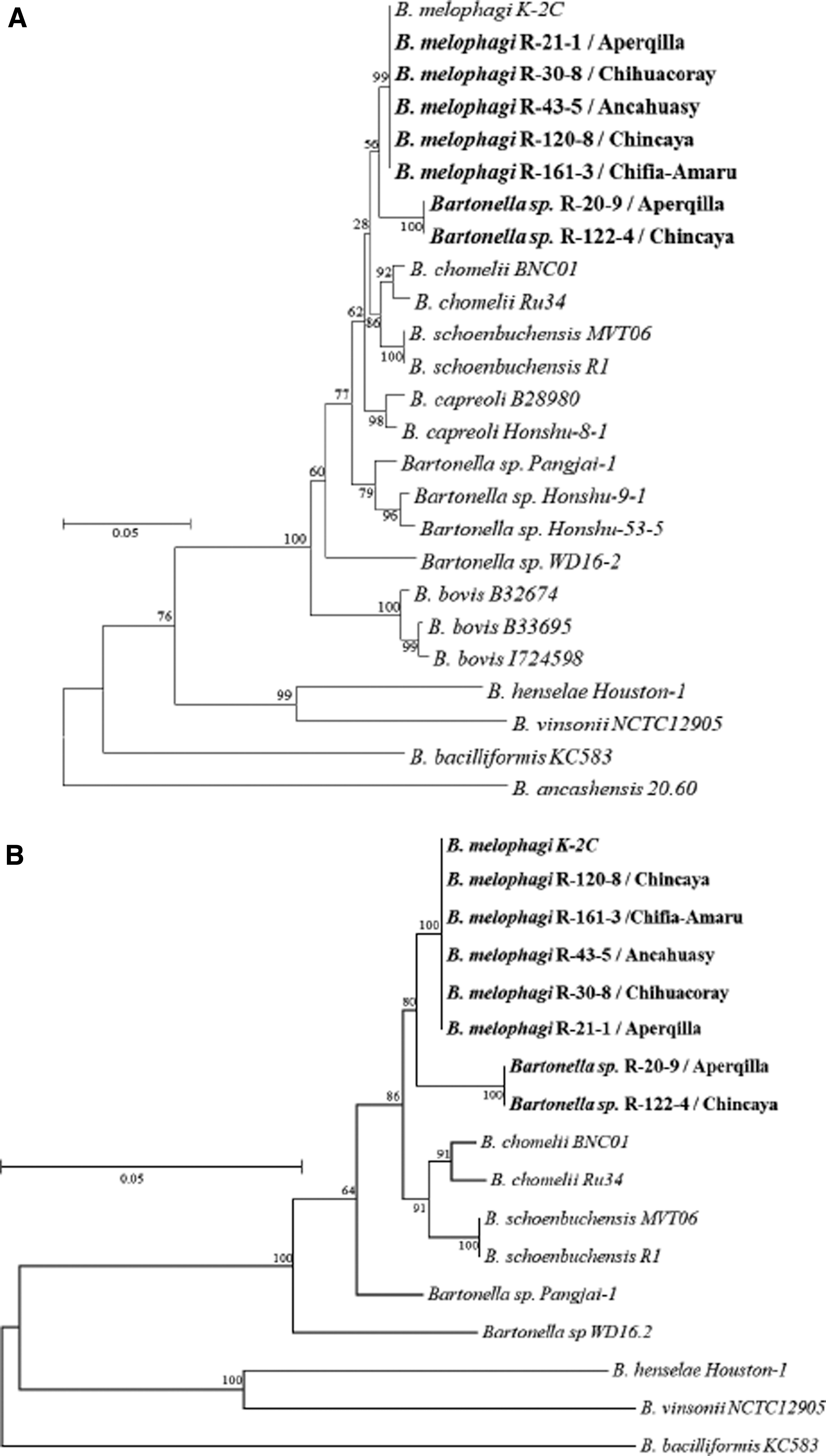
Phylogenetic analysis of seven Bartonella species DNA from ectoparasites collected from domestic animals in Cuzco and known Bartonella species using three and four concatenated gene fragments.
GridION nanopore sequencing output from the eight samples yielded 0.9–2.1 million reads for each library (Table 8). Average read length ranged from 2,310 bases for R-14-3 to 2,913 bases for R-179-9. All libraries had ONT average quality scores >12 except for sample R-14-3 at an average quality score of 10.6. A preliminary analysis of the microbiome was conducted using the online Epi2Me WIMP workflow (
Sequencing Statistics of Eight Sheep Ked Samples Analyzed for Bartonella spp. by WIMP Analysis
Percentage of reads classified as superkingdom (bacteria) or genus (Bartonella) by Epi2Me WIMP workflow.
Seven samples had high numbers of reads matched to the Bartonella genus (43,000–1.2 million reads/sample) that were spread over several species but primarily as Bartonella spp. WD16.2 ID1933904 (33,214–1,034,471 reads per sample). Initial assembly of one of these datasets using the Flye long read assembler and subsequent BLAST analysis of the resulting contigs indicated the species was likely B. melophagi. More detailed examination of the Bartonella species and other microbiome components recovered from these samples will be included in a future publication.
Discussion
The role of domestic livestock ectoparasites in animal and human disease is well known, especially with regard to their direct impact on host well-being (Chomel et al. 2006, Kumsa et al. 2012). This is especially important in consideration of livestock productivity where ectoparasites can affect general health, resulting in weight loss, decreased milk production, and decreased quality of wool and hides. However, knowledge of the role of these arthropods in the transmission of pathogens among livestock or to humans appears to be incomplete as new arthropod-borne diseases associated with new pathogens continue to be discovered in new vectors and/or new locations (Ellis et al. 1999, Parola et al. 2002, Forshey et al. 2010, Kumsa et al. 2012, Flores-Mendoza et al. 2013).
In this study, we surveyed ectoparasites of domestic livestock for the presence of Rickettsia spp. and Bartonella spp. agents in the Department of Cusco, an area known to be endemic for rickettsial diseases such as epidemic typhus (Raoult et al. 1999) and spotted fever group rickettsioses (Schoeler et al. 2005), as well as other arthropod-borne diseases such as bartonellosis (Ellis et al. 1999) and arboviral diseases (Forshey et al. 2010). In the highland regions of Peru, sheep accounted for 80% of domestic livestock. Their wool is a major part of the economy of highland campesinos.
In this field study we surveyed 222 farm animals, 52.7% (117) of them were sheep. The sales of livestock commonly occur in small local fairs. In general, the farm animals freely graze without borders with colorful markings on their ears for identification. Thus, the animals frequently interchange ectoparasites. This maybe one of the main reasons why we found a generalized distribution of genetically similar Bartonella spp. in 14 collections sites. The skin of these domestic animals serves as a reservoir for ectoparasites with the possibility of infection with Bartonella species leading to a zoonosis.
Surprisingly, of 600 ectoparasites investigated in this study, none showed evidence of infection with rickettsiae using standard qPCR-based diagnostics. The arthropods collected included various species of biting/chewing and sucking lice, all of which are found worldwide (Price and Graham 1997). Bovicola ovis, B. caprae, and B. equinus are primarily found in temperate areas where they have not been reported to be infected with rickettsiae or bartonellae (Kumsa et al. 2012).
A study of sucking lice in Hungary found H. eurysternus (1 of 8 pools) to be positive for Rickettsia spp., although at a Ct value of >36 cycles no sequence or molecular confirmation was performed (Hornok et al. 2010). H. suis was negative for rickettsial DNA (Hornok et al. 2010, Flores-Mendoza et al. 2013), but in Algeria, H. suis (7 of 92, 7.61%) were found positive for Rickettsia slovaca (Zeroual et al. 2018). L. stenopsis collected in Hungary were also found to be positive for Rickettsia spp. One of 34 pools was reported as positive for Rickettsia helvetica with a high Ct value of 36.69 (Hornock et al. 2010). The soft ticks O. megnini was negative for Rickettsia spp. (Flores-Mendoza et al. 2013), but there is a report of infection with Babesia in Sri Lanka (Diyes et al. 2018). We did not find rickettsiae or bartonellae in T. penetrans but in Congo it was found they were infested with Candidatus Bartonella rochalimae (Sackal et al. 2008).
The hematophagous ectoparasite of sheep M. ovinus, common name “sheep ked” was the most commonly collected ectoparasite in this study owing to the large number of sheep and donkeys surveyed. Nöller (1917) initially reported M. ovinus infested by Rickettsia melophagi. Later this agent was reclassified as Wolbachia melophagi, and then subsequently determined not to be a Wolbachia spp., but a Bartonella spp., and is considered now as an endosymbiont of sheep keds (Dumler et al. 2001, Halos et al. 2004). Based on DNA sequence data, candidate status was proposed for the new species B. melophagi (M. Vayssier-Taussat, L. Halos, and H.J. Bouluis, unpublished data, available from
Other investigations of sheep keds found no rickettsiae in 22 M. ovinus obtained from Ethiopian sheep (Kumsa et al. 2012), but Hornok et al. (2010) reported 1 of 60 individual M. ovinus was qPCR positive for rickettsiae. However, in this case, the Ct value was 35, and no other molecular confirmation was performed for Rickettsia spp. identification. We detected DNA of B. melophagi agent in M. ovinus in Peru; this agent was also detected in sheep blood in the United States (Bemis and Kania 2007, Kumsa et al. 2014, Kosoy et al. 2016). In addition, sheep keds have also been found to be infected with Rickettsia raoultii and R. slovaca, and two Bartonella species (Bartonella schoenbuchensis and B. chomeli) (Liu et al. 2016), Anaplasma ovis (Zhao et al. 2018), and Trypanosoma (Megatrypanum) melophagium (Costa et al. 1983).
Previous studies found that sequences of fragments of gltA and rpoB genes have the highest discrimination power for Bartonella species identification. Moreover, they proposed that a new Bartonella species would be valid if the 327-bp gltA and 825-bp rpoB fragments shared <96.0% and <95.4% sequence similarity, respectively, between the new agent and recognized Bartonella species (La Scola et al. 2003). Of interest, in this study we found samples (R-20-9 and R-122-4) classified as divergent from B. melophagi strain K-2C owing to their homology to the closest well-characterized species in the phylogenetic analysis. We failed to identify a cluster or distribution patterns among sequenced samples. This suggests that all strains are widely distributed in the study locations.
Conclusions
Sheep keds were the most abundant ectoparasite found infesting sheep and were frequently infected by Bartonella spp. Bartonella species are potential health threats for civilian and military populations.
Bartonella spp. were detected with high infection rates in the sheep ked (52.32%). Two strains were recognized as discriminated by their gene fragment sequences of five genes (gltA, ftsZ, rss, groEL and rpoB). One strain was confirmed to be B. melophagi (100% identity for 4 genes) and one strain as a variant of B. melophagi that only was discriminated by rpoB gene (94.48% identity). M. ovinus and its symbiont B. melophagi are widely spread throughout the New and Old Worlds, however at this point, it is unclear the role B. melophagi may have in human and domestic animal health in Peru. In this study, we determined that Bartonella species, but not Rickettsia species, are widespread among sheep keds of common livestock in the Highlands of Peru.
Footnotes
Acknowledgments
The authors thank the Dirección de Salud, Gobierno Regional de Cuzco, Perú for permission to conduct these studies. The authors also thank Marisa Lozano, Biologist at NAMRU-6 Entomology laboratory, for technical assistance. The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Department of the Navy, Department of Defense, nor the United States.
The experiments reported herein were conducted in compliance with the Animal Welfare Act and in accordance with principles set forth in the “Guide for the Care and Use of Laboratory Animals,” Institute of Laboratory Animals Resources, National Research Council, National Academy Press, 2011. The study protocol was reviewed and approved by the NAMRU-6 Institutional Animal Care and Use Committee in compliance with all applicable federal regulations governing the protection of animals and research.
Some authors were military service members or employees of the U.S. Government during the performance of this study. This work was prepared as part of their official duties. Title 17 U.S.C. §105 provides that “Copyright protection under this title is not available for any work of the United States Government.” Title 17 U.S.C. §101 defines a U.S. Government work as a work prepared by a military service member or employee of the U.S. Government as part of that person's official duties.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
Funding was provided by the Armed Forces Health Surveillance Division Global Emerging Infections Surveillance Branch (GEIS), ProMIS ID P0143_19_N6_04 and USDA National Program 104-Medical Urban and Veterinary Entomology.