
Abstract
Small Indian mongooses (SIMs, Herpestes auropunctatus) have invasively inhabited over 60 islands worldwide. They have been confirmed as a reservoir of rabies, leptospirosis, and salmonellosis; however, their role in the epidemiology of other zoonoses is little known. On St. Kitts, as well as other islands, SIMs harbor Ctenocephalides felis, which can vector several zoonotic diseases. In this study, SIMs were examined for fleas, and the collected fleas analyzed by PCR and DNA sequencing for Bartonella henselae, Rickettsia felis, Yersinia pestis, and Dipylidium caninum. Of the 87 SIMs, 75 (86.2%) harbored C. felis. C. felis recovered from nine (10.3%), one (1.1%), and one (1.1%) of the SIMs was positive for B. henselae, R. felis, and D. caninum, respectively. These data indicate that SIMs serve as an additional reservoir of B. henselae and R. felis, which should be taken into consideration in control and prevention of these rapidly emerging zoonoses.
Introduction
Small Indian mongooses (SIMs, Herpestes auropunctatus) are an invasive species on St. Kitts, a slightly <70 square mile island in the Lesser Antilles in the Caribbean (17°15′N, 62°45′W). Originally introduced to control rats in sugar plantations in the West Indies, SIM is now listed as one of the world's 100 worst invasive species due to its devastating effect on native mammals, birds, reptiles, and amphibians, to the point of extinction in some cases (Barun et al. 2011, Zieger et al. 2014). Natively distributed in Iraq, Iran, Pakistan, Northern India, Nepal, Bangladesh, and Myanmar, SIMs have now been found on over 60 islands worldwide and are spreading to the European continent with populations found in Croatia, Bosnia–Herzegovina, and Montenegro. SIMs are predicted to spread north to Eastern Europe by 2050 due to global climate change (Louppe et al. 2020). In the Caribbean, SIMs have been confirmed as an important reservoir of rabies in Cuba, Granada, and Puerto Rico (Nadin-Davis et al. 2006, 2008, Zieger et al. 2014, Berentsen et al. 2015), leptospirosis in Grenada, St. Kitts, and Trinidad (Everard et al. 1976, Shiokawa et al. 2019), and salmonellosis in Grenada (Miller et al. 2015).
Ctenocephalides felis, the cat flea as it is commonly called, has been found on 138 of 685 (20%) wildlife mammal species on all continents except the Antarctic, including SIMs (Clark et al. 2018). In the Caribbean, SIMs infested with C. felis have been detected on Antigua, Grenada, St. Croix, St. John, St. Kitts, and Trinidad (Nellis and Everard 1983, Cheng et al. 2018). C. felis has been determined to be a capable vector of several pathogens of zoonotic potential, including the bacteria Bartonella henselae, Rickettsia felis, and Yersinia pestis, and it is the intermediate host of the tapeworm, Dipylidium caninum, which has demonstrated zoonotic potential (Jiang et al. 2017).
B. henselae is the causative agent of cat-scratch disease (CSD). CSD is often transmitted from an infected cat to humans by biting or scratching (Klotz et al. 2011). C. felis is a vector for B. henselae transmission between cats, and it may also pass the bacterium to humans through bites (Zangwill et al. 1993, Mosbacher et al. 2011). R. felis is the bacterial agent causing the flea-borne spotted fever. Its primary vector is C. felis, which also serves as a reservoir (Brown and Macaluso 2016). How C. felis transmits R. felis to cats and humans remains debatable, although it very likely occurs when the flea bites the host or through infectious feces deposited by the flea on the host (Legendre and Macaluso 2017). Y. pestis is the bacterial agent causing plague, often occurring in small mammals, which may be spilled over to humans. Transmission between vertebrates occurs by a flea vector or through direct contact with bodily fluids, inhalation of infectious droplets, consumption of carcasses of an infected mammal, or through environmental sources such as bacterium-contaminated soil (Hinnebusch and Schwan 1993, Richgels et al. 2016). D. caninum is a common tapeworm of dogs and cats worldwide. In dogs and cats, which host the adult tapeworm in the small intestine, it typically is not clinically significant. Dogs, cats, and, rarely, people become infected by consuming a flea with the larval stage of D. caninum (Labuschagne et al. 2018). A commonality of these four zoonotic pathogens is that they can be transmitted among mammals, including to humans by the cat flea, C. felis; however, there is a paucity of information on the cat flea's role in transmission of these diseases and specifically in regards to C. felis infesting SIMs.
The aims of the study presented here were to determine if these pathogens occurred in C. felis harbored by SIMs using PCR and DNA sequencing and to analyze phylogenies of the pathogens. Such studies are pivotal in pathogen detection, reservoir determination, and epidemiology of ever-increasing emerging infectious diseases throughout the world.
Materials and Methods
Flea collection, identification, and DNA extraction
All fleas used in the current study were collected from SIMs that were trapped and euthanized from two different locations on St. Kitts and were identified as C. felis as previously determined in the study by Cheng et al. (2018). Specifically, they were identified using a Binocular Stereozoom Microscope (Cole Parmer, Vernon, IL) by morphologic keys of Pratt and Stark (1973). All C. felis collected from each individual SIM were stored in a single container with 70% ethanol and, after identification and photographing, were stored at ∼4°C until DNA extraction. A photograph of each flea was taken using a 5 × objective using a Nikon microscope (Nikon Instruments, Inc., NY) equipped with an Olympus DP27 digital camera (Olympus Corporation of the Americas, PA). The picture was used for sex determination by two of the coauthors C.Y. and J.K. A flea's sex was confirmed as male or female if both concluded the same sex. In contrast, sex was undetermined if they had different opinions, or at least one was unsure about its sex.
Each flea was measured in body weight (μg) using a Sartorius Lab Balance CPA225D (Sartorius, Goettingen, Germany) after being rinsed thrice in 500 μL phosphate buffered saline (PBS) by vortex for 10 s followed by being gently blotted dry with a Kimwipe tissue (KIMTECH, Roswell, GA). Afterward, each individual flea was ground in a 1.5 mL microcentrifuge tube in 25 μL PBS using a PELLET PESTLE® Cordless Motor homogenizer (Kimble Chase Life Science and Research Products, LLC, Vineland, NJ) until no “chunks” were seen. DNA was then extracted using QIAGEN DNeasy Blood and Tissue Kit (QIAGEN, Hilden, Germany) as per the manufacturer's instructions and eluted in 100 μL elution buffer. DNA quality and quantity were monitored using NanoPhotometer (Implen, Inc., Westlake Village, CA). Extracted DNA was stored at −20°C.
Strategy
The study strategy is outlined in the flow chart in Fig. 1. First, the extracted flea DNA was assessed for its suitability for PCR amplification of the 18S rRNA gene of C. felis. Extracted DNA that was PCR negative for C. felis was deemed unsuitable for further testing, and the fleas from which the DNA had been extracted were excluded in calculating prevalence of the pathogens of interest, namely B. henselae, R. felis, Y. pestis bacteria, and D. caninum. Next, the 18S rRNA-positive flea DNA samples were individually tested by a multiplex PCR for all four pathogens using six pairs of primers (Supplementary Table S1). Afterward, purified PCR amplicon of simplex PCR using positive multiplex PCR mixtures as templates was subjected to DNA sequencing in both directions using PCR primers. The DNA sequence was then used in BLAST search against the nucleotides deposited in National Center for Biotechnology Information. Finally, a phylogenetic analysis was performed.
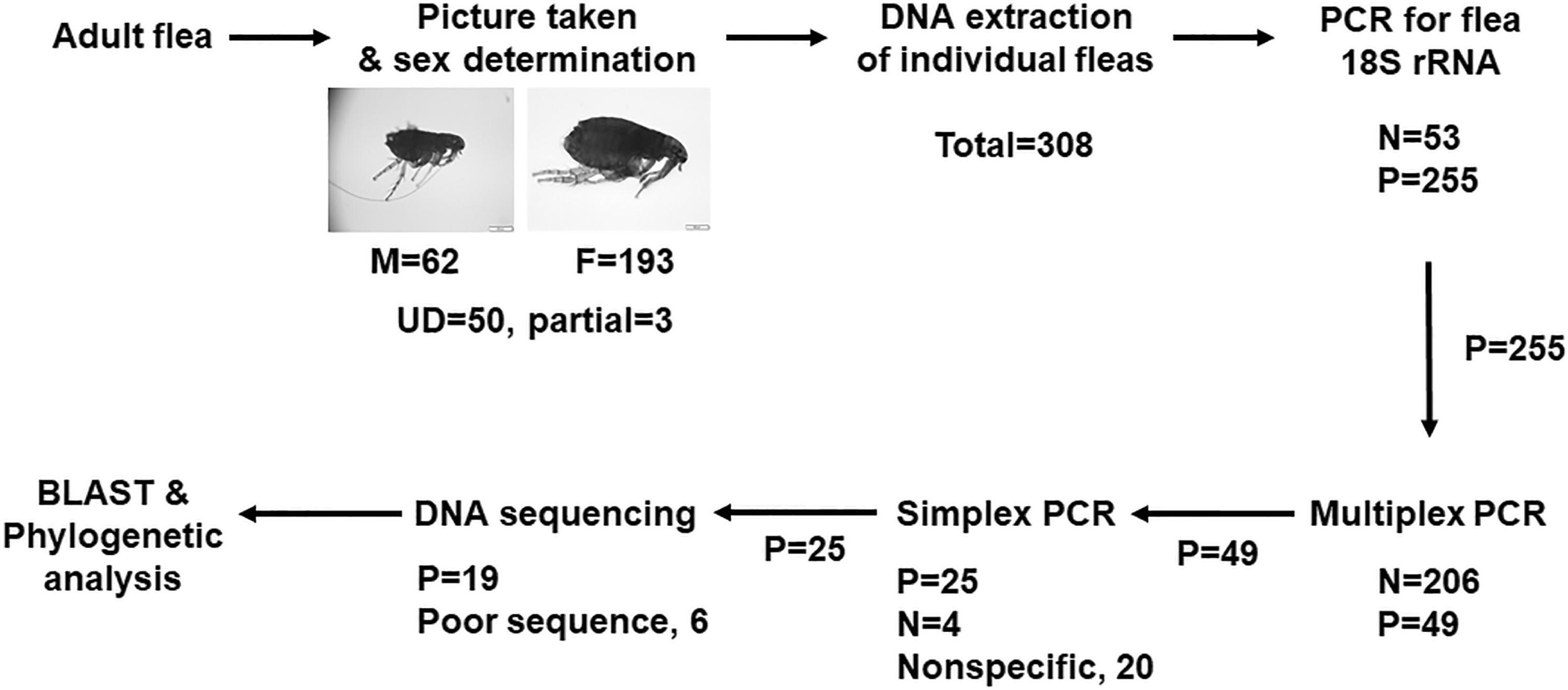
Flow chart of the strategy taken in the current study. A multiplex PCR detected DNAs of four zoonotic pathogens, including Bartonella henselae, Rickettsia felis, Yersinia pestis, and Dipylidium caninum. UD, undetermined.
PCR and DNA sequencing
The primers selected were based on previous publications and included: URBarto1 and URBarto2 (expected size 681 bp) and CAT1 and CAT2 (418 bp) for B. henselae tRNA and serine protease (htrA) gene, respectively (Rolain et al. 2003, Mazaheri Nezhad Fard et al. 2016); BM59 and B807 (862 bp), CS-877 and CS-1273 (382 bp) for R. felis outer membrane protein B (OmpB) and citrate synthase (gltA) genes, respectively (Roux and Raoult 2000, Rolain et al. 2003); Yp1 and Yp2 (480 bp) for Y. pestis plasminogen activator (pla) gene (Hinnebusch and Schwan 1993); DS28S-1F and DS28S-1R (653 bp) for D. caninum 28S rRNA gene (Beugnet et al. 2014); and 18S a1.0 and 18S 9R (980 bp) for C. felis 18S rRNA gene (Hornok et al. 2018). All primers were synthesized by Integrated DNA Technologies (Coralville, IA). The sequence and references for all primers are listed in Supplementary Table S1.
The primers 18S a1.0 and 18S 9R targeted a fragment of sequences shared by members of seven flea genera, including cat flea (Hornok et al. 2018). Flea DNA was first used to amplify the C. felis 18S rRNA in HotStart Taq Plus Master Mix (QIAGEN) in a final volume of 25 μL, including 10 μL DNA, and 1 μM each primer of 18S a1.0 and 18S 9R. PCR was performed in the Mastercycler Nexus Gradient thermal cycler (Eppendorf, Enfield, CT) with the following thermal cycle: 95°C 2 min, 35 cycles of 95°C 30 s, 55°C 1 min, and 72°C 2 min followed by a final extension of 72°C 10 min. PCR products were detected by electrophoresis in 1% agarose with 100 bp DNA ladder (Invitrogen, San Francisco, CA). The 18S rRNA positive DNA samples were then individually subjected to a multiplex PCR. The latter was performed in 50 μL reaction, including 0.5 μM of each primer, 0.2 mM dNTPs, 2.5 U Taq DNA polymerase (TaKaRa, Clontech, Mountain View, CA), and 30 μL of individual flea DNA. The thermal cycle was the same as outlined above except that the annealing temperature was 52°C. In total, six pairs of primers were used in each multiplex PCR. One half of PCR mix was subjected to electrophoresis as just described. The remaining half was used as a template for simplex PCRs, in which 1 μL of the positive multiplex PCR mix was used in a 20 μL reaction mix. Only one pair of primer was used; therefore, for each positive multiplex PCR, six simplex PCRs were performed. The same reagents and thermal cycles as those of PCR for the flea 18S rRNA were used. Gel electrophoresis with all 20 μL PCR mix was used to detect PCR amplicons. For the positive simplex PCR, a repeated simplex PCR under the same conditions with 50 μL PCR mixture was performed to generate enough amplicon for DNA sequencing.
The amplicons of simplex PCR were purified using QIAquick Gel Extraction Kit (QIAGEN) after electrophoresis or directly extracted with the QIAquick PCR Purification Kit (QIAGEN). The purified DNA was analyzed by both NanoPhotometer (Implen, Inc.) and gel electrophoresis and was directly sequenced in both directions using the PCR primers (Macrogen, Seoul, Korea).
Phylogenetic analysis
Phylogenetic analysis was performed using Molecular Evolutionary Genetics Analysis (MEGA) software, version 7 (Kumar et al. 2016) using both the Maximum Likelihood method-Tamura-Nei model (Tamura and Nei 1993) and Neighbor-joining-Maximum Composite Likelihood (Saitou and Nei 1987) with 1000 bootstrap replications. The default setting of MEGA 7.0 was used for both analyses.
Results
Among 87 mongooses examined, 75 (86.21%) were found infested with at least one cat flea (Table 1). This was slightly higher than our earlier report of 79.3% (Cheng et al. 2018). The number of fleas collected from each mongoose ranged from 1 to 14 with an average of 4.3 and a median of 3.
Prevalence of Bartonella henselae, Rickettsia felis, Yersinia pestis, and Dipylidium caninum Among Small Indian Mongooses and Ctenocephalides felis
Among 308 fleas examined, 193 were females, 62 were males, and for 53 the sex could not be confirmed (Table 1). Among 308 individual flea DNA extractions, 255 DNA samples (82.79%) tested positive, whereas 53 (17.21%) were negative by PCR amplification for the C. felis 18S rRNA gene. After multiplex PCR amplification, 49 of these 255 DNA samples were positive with at least one PCR amplicon. It was determined by simplex PCR and DNA sequencing that 19 of these 49 yielded correct DNA fragments and sequences (Fig. 1).
The prevalence of SIMs with C. felis positive for B. henselae, R. felis, D. caninum, and Y. pestis was 10.34%, 1.15%, 1.15%, and 0%, respectively (Table 1). The prevalence for the pathogens examined in C. felis was: B. henselae—5.88% (15/255), R. felis—0.39% (1/255), D. caninum—0.39% (1/255), and Y. pestis—0% (0/255) (Table 1). Interestingly, not a single male flea was found positive with any of these pathogens (Table 2). Among the 16 fleas found positive for the zoonotic pathogens, all except one was infected with only a single pathogen of interest. The one exception was coinfected with B. henselae and R. felis (Table 2). All the DNA sequences reported in this study are available from GenBank with the accession numbers listed in Table 2.
Zoonotic Pathogens Detected in Ctenocephalides felis on Small Indian Mongooses by PCR and DNA Sequencing
htrA, serine protease; gltA, citrate synthase; ompB, outer membrane protein B; UD, undetermined.
B. henselae infection of C. felis
With the two pairs of primers used to detect B. henselae, htrA and tRNA, 15 fleas and 1 flea were found positive, respectively. After DNA sequencing, only one single nucleotide polymorphism was found among the 15 htrA sequences, that is, a “G” in 12 of them was replaced by an “A” in the remaining three (Supplementary Fig. S1). Nevertheless, this replacement resulted in no changes in amino acid sequence (Supplementary Fig. S2). Therefore, the function of this protein should be the same for all 15 isolates. Phylogenetic analysis using B. quintana as an outgroup showed that these 15 new sequences formed a clade with those reported from different geographic locations throughout the world originating from such hosts as humans, cats, and even ticks. However, a sequence (Accession # DQ874333) derived from an Iranian cat designated as B. henselae was separated from all B. henselae and even B. quintana. This was almost certainly a different Bartonella sp. (Fig. 2A), which should be further explored.

Molecular phylogenetic analysis of B. henselae in infected Ctenocephalides felis on small Indian mongooses. The methods applied were the Maximum Likelihood method using the Tamura-Nei model and Neighbor-joining-Maximum Composite Likelihood. Both methods yielded similar results. Only data by the Maximum Likelihood method are presented. Each entry is presented in the order of bacterium name, host, location, and GenBank accession number, if known. The analysis was performed with 1000 bootstrap replications. The new DNA sequences described in the current study are marked by “•”. Scale shows the number of substitutions per site.
Among the 15 fleas that tested positive for B. henselae htrA gene, only one was PCR positive when the tRNA fragment was targeted. BLAST search determined that it had the highest identify to AB674235, a DNA sequence designated as Bartonella sp. and originating from the whole blood of a Japanese badger, Meles anakuma (Sato et al. 2012). Further analysis of alignment of the two DNA sequences showed that the new one, MT048286, was 32 bp shorter than AB674235. The identity between them was only 89.14% (583/654) (Supplementary Fig. S3). It was not surprising that these two, along with another sequence with accession number of EU098132, formed a clade in a phylogenetic analysis (Fig. 2B). Currently there are no homologous sequences that are labeled as B. henselae, although some are identified to be B. clarridgeiae and B. rudakovii (Fig. 2B). Consequently, both BLAST and phylogenetic analysis failed to determine this new DNA sequence as B. henselae. However, it would be reasonable to extrapolate it as such provided (1) that the same flea tested positive for B. henselae when PCR and DNA sequencing targeted the htrA gene; (2) that all 15 htrA sequences were confirmed as B. henselae; and (3) 26.03% (38/146) and 13.70% (20/146) of cats on St. Kitts in 2012 were found positive for B. henselae and B. clarridgeiae by PCR and DNA sequencing of Bartonella spp. gltA gene in the whole blood (Huang et al. 2019). Consequently, all three sequences with accession numbers MT048286, AB674235, and EU098132 are considered B. henselae. Collectively, PCR and DNA sequencing of both htrA and tRNA identified B. henselae infected cat fleas collected from SIMs on St. Kitts.
R. felis infection of C. felis
To determine prevalence of R. felis among cat fleas, two pairs of primers targeting the OmpB and gltA genes were used in PCR. Only a single flea tested positive for both genes. The new OmpB sequence described here was 99.64% identical to those of R. felis with the accession numbers KF056801 and CP000053 deposited in GenBank. Similarly, the new gltA sequence was 99.48% identical to one that has already been published with a GenBank accession number of GQ329873 (Behar et al. 2010). In contrast, the new sequences shared only 92.46% and 95.81% identity of those of R. asembonensis corresponding genes with accession numbers KY650699 and MN003395, respectively. Phylogenetic analysis of both genes showed that R. felis formed two clades (Fig. 3). The same flea was also positive for B. henselae (Table 2).

Molecular phylogenetic analysis of R. felis in infected C. felis on small Indian mongooses by Maximum Likelihood method using the Tamura-Nei model. Neighbor-joining-Maximum Composite Likelihood yielded similar results that are not presented. Each entry is presented in the order of bacterium name, host, location, and GenBank accession number, if known. The analysis was performed with 1000 bootstrap replications. The new DNA sequences described in the current study are marked by “•”. Scale shows the number of substitutions per site.
D. caninum infection of C. felis
The 28S rRNA fragment of D. caninum was targeted in PCR. Only one flea was found positive with D. caninum DNA. The new DNA sequence with an accession number of MT048289 bore 97.62% (614/629) identity to a DNA sequence in GenBank (MH182478). Phylogenetic analysis of this new DNA sequence along with those available entries in GenBank showed three distinct clades, named I, II, and III (Fig. 4). Interestingly, this new sequence was by itself in clade III. All three clades of D. caninum were distinctly separated from out-groups of Glossocercus caribaensis and Dilepis undula (Fig. 4).
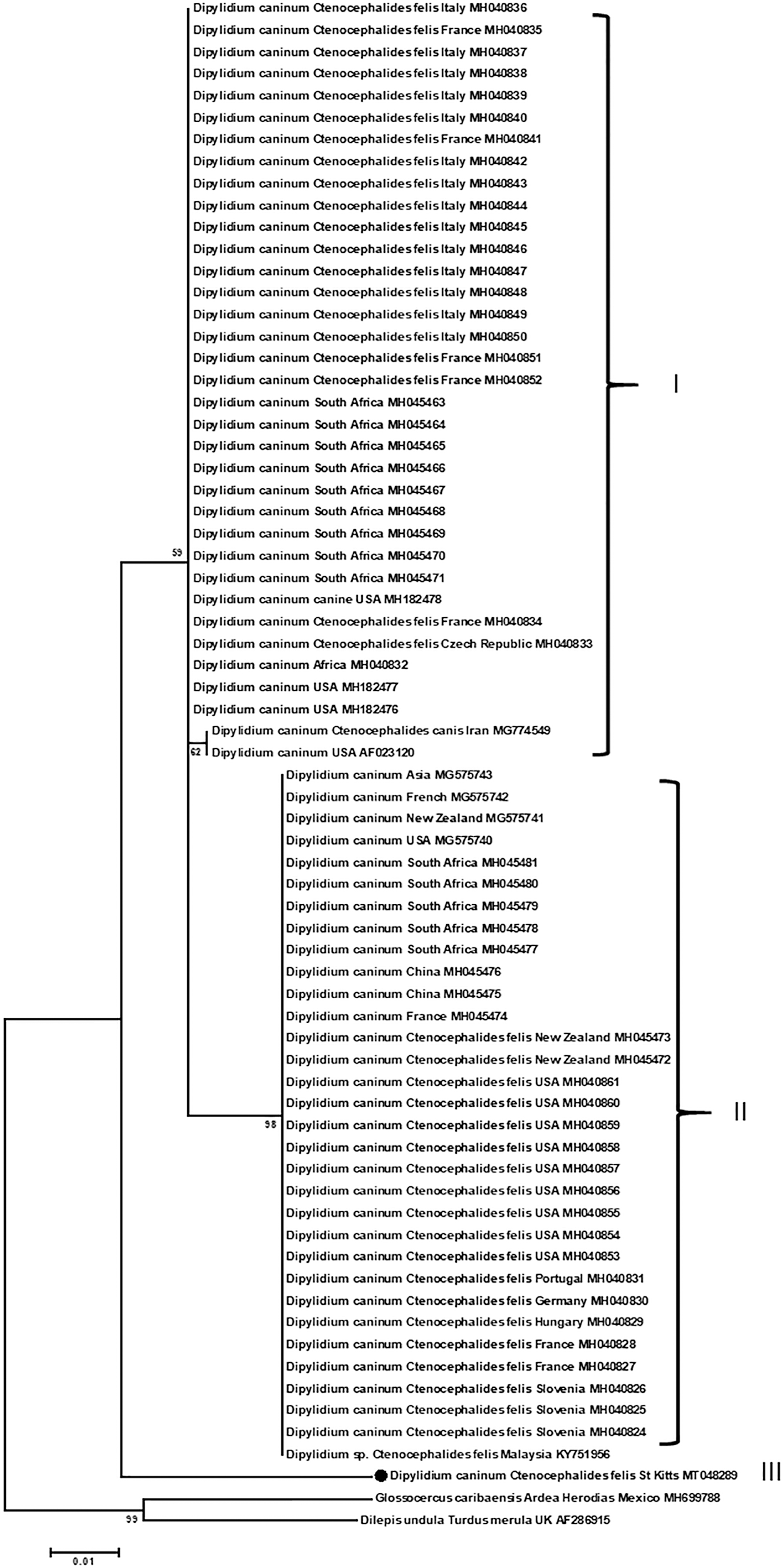
Molecular phylogenetic analysis of 28S rRNA of D. caninum in infected C. felis on small Indian mongooses by Maximum Likelihood method using the Tamura-Nei model. Similar results were obtained by Neighbor-joining-Maximum Composite Likelihood and are not presented. Each entry is presented in the order of parasite name, host, location, and GenBank accession number, if known. The analysis was performed with 1000 bootstrap replications. The new DNA sequences described in the current study are marked by “•”. Scale shows the number of substitutions per site. Clade I and II are the canine and feline strains, respectively, as previously described (Labuschagne et al. 2018). Glossocercus caribaensis and Dilepis undula are outgroups.
Discussion
In this study, individual cat fleas harbored by SIMs on the Caribbean island of St. Kitts were tested by PCR and DNA sequencing for four zoonotic pathogens, B. henselae, R. felis, and Y. pestis bacteria and D. caninum tapeworm. To the best of our knowledge this is the first study to examine individual C. felis of SIMs for zoonotic pathogens. An advantage of testing individual fleas is that prevalence of these pathogens among C. felis populations can be accurately determined. An obstacle, however, is that limited amounts of DNA are available from each flea. Our strategy was to use multiplex PCR followed by simplex PCR. Multiplex PCR was designed to amplify all four zoonotic pathogens targeting six different genes, that is, two targets each for B. henselae and R. felis and one each for Y. pestis and D. caninum, with various sizes of expected amplicons. This strategy worked satisfactorily, leading to the detection of three of four pathogens with the prevalence of cat fleas with positive pathogens ranging from 0.39% to 5.88% and that of SIMs with fleas with positive pathogens from 1.15% to 10.34%. Ehlers et al. (2020) tested pooled fleas collected from various animals, including Grandidier's mongooses, Galidictis grandidieri, on Madagascar for rickettsiae, borreliae, bartonellae, and Y. pestis. They only found Rickettsia sp. in Echidnophaga gallinacean on mongooses. Due to pooled samples from various hosts, the prevalence of rickettsiae in the E. gallinacean could not be confirmed although a minimal infection rate of 8% was extrapolated (Ehlers et al. 2020).
In a survey of SIMs on the Caribbean island of Grenada, West Indies, 32.3% (54/167) were found seropositive with antibodies to B. henselae and 35.3% (18/53) of their blood contained B. henselae DNA by PCR detection of gltA and/or the β subunit of RNA polymerase (rpoB) genes (Jaffe et al. 2018). Previously, 15.9% (10/63) of clinically normal SIMs were found positive for B. henselae by blood culture in Japan (Sato et al. 2013). In the study presented here, 5.88% (15/255) of the cat fleas were found positive with B. henselae DNA by conventional PCR and DNA sequencing targeting htrA gene. However, only one of 15 htrA-positive fleas was also positive by tRNA, which suggests that conventional PCR targeting tRNA fragment of this bacterium is much less sensitive than that amplifying the htrA gene. These 15 fleas were found on 9 of the 87 SIMs (10.34%). Two or more B. henselae DNA positive fleas were found on each of four SIMs, indicating that they had obtained the pathogen from the SIMs (Table 3). This highlights the SIMs as a potential source of the flea's B. henselae even in the absence of direct evidence of the bacterial DNA in the SIMs. Therefore, SIMs are a reservoir of B. henselae.
Positive Rate of Ctenocephalides felis for Bartonella henselae DNA Among Small Indian Mongooses Harboring More Than One Flea and Having at Least One Positive Flea
In the study presented here, 1 of 255 cat fleas was confirmed positive with R. felis DNA by PCR and DNA sequencing of two targeted genes, gltA and ompB. Previously, 11 of 57 C. felis (19.30%) collected from cats on St. Kitts tested positive for R. felis targeting ompA or gltA. Furthermore, on Dominica, 1 of 32 C. felis (3.13%) on cats was positive for R. felis ompA (Kelly et al. 2010). In a survey of cats using PCR detecting R. felis DNA for the same gene in the whole blood, none of 119 cats (68 in 2011 and 51 in 2014) was found positive. Nevertheless, 42% (22/52 in 2014) of cats were found positive with antibodies to spotted fever group Rickettsia by indirect fluorescence assay (Kelly et al. 2017). Collectively, SIMs along with cat fleas are reservoirs of R. felis.
D. caninum is a tapeworm commonly found in domestic dogs and cats. It is occasionally diagnosed in humans, especially in young children (Jiang et al. 2017, Hogan and Schwenk 2019, Portokalidou et al. 2019). It has an indirect life cycle. A definitive host becomes infected by ingestion of the cysticercoid larvae in an intermediate host such as an adult flea (Beugnet et al. 2014, Labuschagne et al. 2018). The PCR positive rate of cat fleas collected from SIMs in the current study was 0.39%, which was much lower than those collected from cats (2.23%; 44/1969) and from dogs (5.20%; 38/732) from several European countries (Beugnet et al. 2014). It has interestingly been found that Dipylidium sp. collected from dogs was different from cats and very likely at the species level. This difference further extends to the Dipylidium-positive cat flea, C. felis, harvested from dogs or cats (Labuschagne et al. 2018). The phylogenetic analysis of the newly discovered 28S rRNA sequence in the study presented here with fleas from SIMs was distinctly separated from the clades of canine and feline isolates (Fig. 4). Further study using adult specimens and infected cat flea samples from cats, dogs, and mongooses on St. Kitts is warranted to confirm that this could represent another Dipylidium species.
Among 255 cat fleas tested for Y. pestis, none was found positive. This negative finding indicates that SIMs are unlikely hosts of Y. pestis or Y. pestis is nonendemic on St. Kitts. Alternatively, it may suggest that C. felis is not a natural vector for Y. pestis. In a study testing pooled fleas for Yersinia sp. in Madagascar, 3 of 46 pools of E. gallinacean (n = 289) and 1 of 16 pools of Xenopsylla cheopis (n = 59) were found positive. In contrast, none of six pools of C. felis (n = 31) and none of four pools of Pulex irritans (n = 10) were positive (Ehlers et al. 2020). In a study mimicking natural blood feeding in the laboratory, C. felis was found with limited vector capacity for Y. pestis (Bland and Hinnebusch 2016).
Conclusions
Individual C. felis fleas infesting SIMs on St. Kitts, West Indies were tested for four zoonotic pathogens using multiplex and simplex PCR followed by DNA sequencing. These fleas were found positive for B. henselae, R. felis, and D. caninum. SIMs are a new reservoir of widely distributed B. henselae and R. felis.
Ethics Approval and Consent to Participate
Animal use for this study was approved (approval #15-2-007) by the Institutional Animal Care and Use Committee (IACUC) of Ross University School of Veterinary Medicine. All animals were used and cared for in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the United States National Institutes of Health.
Footnotes
Acknowledgments
The authors are very grateful to Aria Armendariz Peavy, Francesca DeMasi DiFroscia, Ani Sefilzhian, Staci Barrow, and Kristen Erickson of Ross University School of Veterinary Medicine for their help in taking flea pictures, extracting flea DNA, and/or performing PCR.
Authors' Contributions
C.Y. and A.D. conceived the study. K.F., K.P., and X.C. performed experiments. C.Y. and J.K. obtained the funding and performed flea sex determination. C.Y. managed the study and DNA sequencing, performed phylogenetic analysis, and wrote the article. All authors read and approved the final version of the article.
Disclaimer
The funding source played no role in the design of the study and collection, analysis, and interpretation of data and in writing the article.
Author Disclosure Statement
No conflicting financial interests exist.
Funding Information
The study was partially supported by an intramural grant of Ross University School of Veterinary Medicine (Vector-borne pathogens, C.Y. and J.K.). The page charge was paid by the Center One of Ross University School of Veterinary Medicine.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.