
Abstract
The circulation of arboviruses in livestock ruminants has often gone unrecognized owing to the fact that a significant percentage of arboviruses probably induce subclinical infections and/or negligible symptoms in infected animals. To determine the current situation of arbovirus circulation in the Yaeyama Islands, attempts to isolate viruses from bovine blood samples collected between 2014 and 2019 have been made. In total, 308 blood samples were collected during the study period, and 43 of them induced cytopathic effects (CPEs) in cell cultures. The identification of the CPE agents was performed by reported RT-PCR assays and a high-throughput analysis with a next-generation sequencing platform. The obtained viruses consisted of an orthobunyavirus (Peaton virus), Culicoides-borne orbiviruses (bluetongue virus serotypes 12 and 16, epizootic hemorrhagic disease virus [EHDV] serotypes 5, 6, and 7, D'Aguilar virus, and Bunyip Creek virus), and potential mosquito-borne orbiviruses (Yunnan orbivirus, Guangxi orbivirus, and Yonaguni orbivirus). Most of the orbiviruses were recovered from washed blood cells with mosquito cell cultures, suggesting that this combination was more efficient than other combinations such as plasma/blood cells and hamster cell lines. This marked the first time that the isolation of EHDV serotypes 5 and 6 and three potential mosquito-borne orbiviruses was recorded in Japan, showing a greater variety of orbiviruses on the islands than previously known. Genetic analysis of the isolated orbiviruses suggested that the Yaeyama Islands and its neighboring regions were epidemiologically related. Some of the viruses, especially the potential mosquito-borne orbiviruses, were isolated during several consecutive years, indicating their establishment on the islands.
Introduction
Arthropod-borne viruses (arboviruses) are transmitted by hematophagous insects and ticks and constitute a diverse taxonomic group of viruses that generally need invertebrate vectors in their natural transmission cycle (Gubler and Vasilakis 2016). Many arboviruses infect humans and domestic animals, and some of them cause severe illnesses in humans and animals. Several arboviruses lead to severe clinical signs in domestic ruminants and subsequent economic loss in livestock industries. For example, bluetongue virus (BTV) and epizootic hemorrhagic disease virus (EHDV) are Culicoides-borne orbiviruses of significant animal health concern owing to their worldwide distribution and association with severe diseases in domestic and wild ruminants (Maclachlan et al. 2019). On the contrary, a significant percentage of arboviruses induce subclinical infections and/or negligible symptoms in their ruminant hosts. Even arboviruses pathogenic to ruminants often have low morbidity in infected hosts. Therefore, the circulation of arboviruses within a population has probably gone unrecognized in many cases.
Sentinel surveillance systems are sensitive detection systems for silent arbovirus circulation (Racloz et al. 2006). These systems are useful for monitoring the prevalence of endemic arboviruses and as tools for the early detection of exotic arboviruses. Virus isolation from sentinel animals independent of clinical signs may reveal the incursion and circulation of new or unexpected arboviruses (Gard et al. 1988a, Kato et al. 2016a). However, the efficacies of each virus isolation system probably should be evaluated, because differences between systems may introduce a strong bias in their surveillance sensitivities. For example, the susceptibilities of cultured cell lines to each arbovirus significantly affect the efficacy of the isolations (Gard et al. 1988a, Wechsler and McHolland 1988, Eschbaumer et al. 2012, Kato et al. 2016a).
Other than BTV and EHDV, several orbiviruses are known to be involved in domestic ruminant diseases. Chuzan virus (CHUV) that is a strain of Kasba virus serotype (species Palyam virus) causes bovine malformations characterized by a hydranencephaly–cerebellar hypoplasia syndrome (Goto et al. 1988). D'Aguilar virus (DAGV), another serotype of the species Palyam virus, was also suggested to be involved in the similar manifestations in calves (Ohashi et al. 2004a). In addition, many novel orbivirus species and new serotypes of known orbivirus species have been identified in infected domestic ruminants without obvious clinical signs since this century began (Sun et al. 2016, Attoui and Jaafar 2015, Shirafuji et al. 2017, Ries et al. 2020). However, their global distribution, ecology, and pathogenesis remain uncertain.
Previous arbovirus surveillance in sentinel cattle was conducted between 1994 and 2014 in the Yaeyama Islands, which are located in the southwesternmost part of Japan (Kato et al. 2016b). During the surveillance, a total of 33 isolates composed of three orthobunyavirus species, three Palyam virus (PALV) serotypes, and an EHDV of which serotype is unidentified were obtained using hamster cell cultures. The islands are in the subtropical zone, and cattle production is flourishing there. Therefore, these islands may be an ideal area for arbovirus maintenance in vertebrate–vector cycles. In fact, an epizootic outbreak of bovine ephemeral fever continued for ∼13 months (2012–2013) in the islands (Hayama et al. 2016). In addition, many ruminant arboviruses have been circulating in Taiwan and the southern part of mainland China (Yanase et al. 2020), suggesting that the islands is a better location to detect arbovirus spreading around Japan.
In this study, we further attempted to isolate arboviruses from sentinel cattle on the Yaeyama Islands between 2014 and 2019 to obtain updated information on the recent situation. In addition to the previously used hamster cell lines, a mosquito-established cell line was used for virus isolation to increase the detection sensitivity for arboviruses. We obtained a variety of Culicoides and mosquito-borne orbiviruses from bovine blood and genetically characterized them by using a next-generation sequencing (NGS) platform and a Sanger sequencing approach.
Materials and Methods
Study area
The Yaeyama Islands consist of 23 islands and are located in the southwestern part (24.00–26.67° N, 122.75–124.50° E) of the Ryukyu archipelago, Japan (Fig. 1). The islands have a humid subtropical climate. The mean temperatures are 17.8–20.5°C in the coldest month (January) and 28.5–30.4°C in the warmest month (July), and the annual precipitation was 979.0–3053.5 mm among Ishigaki, Iriomote, and Yonaguni Islands between 2014 and 2019 (Japan Meteorological Agency). Typhoons often approach in summer and autumn. The prevailing winds are southerly in summer and northerly or easterly in the other seasons. In the Yaeyama Islands, Ishigaki and Iriomote Islands have relatively large area (222.24 and 289.27 km2, respectively). Both islands are covered by laurel forests. Ishigaki Island has been well developed and farmlands and pastures spread out in the southern part of the island. Paddy fields are also scattered through the island. The other islands are small (< 30 km2) and do not have rich vegetation. No wild ruminant species is distributed in the Yaeyama Islands. The cattle population was estimated to be ∼30,000 head during the surveillance period. Most of them (>99%) were beef cattle (Japanese black). The Yaeyama Islands also had ∼1300–2000 goats, 100 water buffalos, and 250–450 horses.
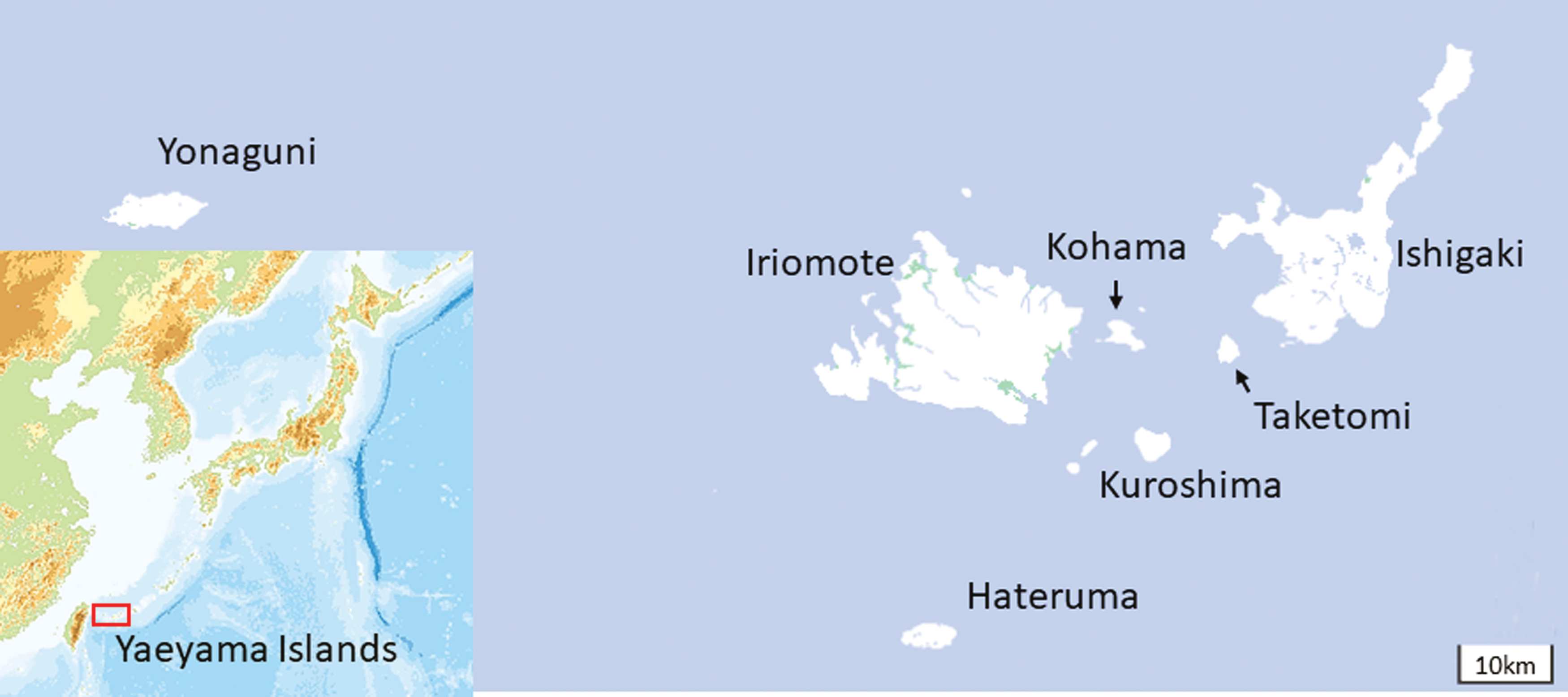
Geographical location of the Yaeyama Islands. The maps were provided by the Geospatial Information Authority of Japan. Color images are available online.
Blood sampling
Heparinized blood samples were collected from 40 to 60 healthy cattle (Japanese black) in late November through early December between 2014 and 2019. The donors were randomly selected from 4- to 12-month-old cattle without movement history. In total, 308 blood samples were obtained on the islands of Ishigaki, Taketomi, Kuroshima, Kohama, Iriomote, Hateruma, and Yonaguni and used for further processing (Table 1). Bovine blood samples were collected in Venoject II vacuum blood collecting tubes containing heparin sodium (Terumo, Tokyo, Japan). After collection, the samples were chilled and transported to the laboratory. The blood samples were separated into plasma and blood cells by centrifugation (2000 g at 4°C for 15 min) with a refrigerated centrifuge (model LX-130; Tomy Tokyo, Japan) and the plasma was obtained. The sedimented blood cells were washed three times with phosphate-buffered saline (PBS) to remove antibodies and the final pellet was suspended in PBS. The plasma and blood cell suspensions were stored at −80°C until use in virus isolation attempts.
Collection Date and Location of Bovine Blood Samples
Virus isolation
The preserved plasma and blood cells were thawed and were inoculated onto monolayer cultures of baby hamster kidney (BHK-21), hamster lung (HmLu-1) and mosquito (C6/36) cells as described previously (Kato et al. 2016a, Murota et al. 2020). In brief, BHK-21 and HmLu-1 cells cultured in test tubes with Eagle's minimum essential medium (MEM; Nissui, Tokyo, Japan) supplemented with 0.295% tryptose phosphate broth (TPB; Becton Dickinson and Company, Franklin Lake, NJ), 0.1% sodium bicarbonate, and 5–10% heat-inactivated bovine serum. The monolayered cell cultures were washed three times with Earl's solution and then 100 μL of the sample was added to the washed cell cultures. The inoculated cells were incubated in maintenance medium (MEM supplemented with 0.295% TPB, 0.15% sodium bicarbonate, and 10 μg/mL gentamicin sulfate) by rotation at 37°C. Monolayers of C6/36 cells were grown in six-well plates with MEM supplemented with 0.295% TPB, 0.1% sodium bicarbonate, and 10% heat-inactivated fetal bovine serum in 5% CO2 overnight and were washed three times with Earl's solution before inoculation. Then, the C6/36 cells were inoculated with 200 μL of each blood cell suspension. In the case of the plasma samples, 50 μL of each was diluted in 200 μL of maintenance medium and applied to the C6/36 monolayer. The inoculated C6/36 cells were incubated in maintenance medium at 28°C in 5% CO2. The inoculated cell cultures were observed for cytopathic effects (CPEs) over 7 days and were passed twice in the same manner if CPE was not observed. The supernatants of cell cultures with CPEs were preserved at −80°C until further characterization.
Virus identification
RNA extraction from the supernatants of infected cell cultures was conducted using the High Pure Viral RNA Kit (Roche Diagnostics, Mannheim, Germany) according to the manufacturer's instructions. Group-specific RT-PCRs targeting the small (S) RNA segment of Simbu serogroup orthobunyaviruses, which encodes the nucleocapsid protein, and segment (Seg) 3 of PALV, EHDV, and BTV were performed as previously described (Ohashi et al. 2004b, Purnomo Edi et al. 2017). If the samples tested positive for the aforementioned virus groups, further identification was attempted through virus/serotype-specific RT-PCRs with previously described primer sets targeting the medium (M) RNA segments of Simbu serogroup orthobunyaviruses, which encodes the viral surface glycoproteins (Purnomo Edi et al. 2017), and Seg2 of EHDV (Maan et al. 2010, Hirashima et al. 2015), BTV (Maan et al. 2012), and PALV (Kato et al. 2016a) (Supplementary Table S1). A primer set for the specific detection of Seg2 of Bunyip Creek virus (BCV) was newly designed in this study (Supplementary Table S1). Direct sequencing of the PCR products was performed with BigDye Terminator v. 3.1 (Thermo Fisher Scientific, Waltham, MA) on an ABI 3100-Avanti Genetic Analyzer (Thermo Fisher Scientific). A homology search was conducted between the obtained sequences and the International Nucleotide Sequence Databases (INSD; DDBJ/EMBL/GenBank) at the nucleotide (nt) (BLASTN) and the amino acid (aa) (BLASTX) levels. The serotype definition of BTV, EHDV, and PALV were also referred to nt variations of Seg2 within the same serotype of each species (Maan et al. 2010, 2016, Ebersohn et al. 2019).
The selected CPE agents that could not be identified by the aforementioned RT-PCRs were subjected to NGS analysis as described previously (Murota et al. 2020). In brief, removal of the host contaminant nucleic acids from the culture supernatants was performed with a nuclease mixture including 0.01 U/μL TURBO™ DNase (Thermo Fisher Scientific), 0.01 U/μL Baseline Zero™ DNase (Lucigen, Middleton, WI), and 0.1 ng/μL RNase solution (Nippon Gene, Tokyo, Japan) for 30 min at 37°C. After the digestion of host contaminants, viral RNAs were extracted by Isogen II (Nippon Gene). Double-stranded cDNA synthesis of the extracted RNAs was conducted with the NEBNext RNA Ultra First Strand Synthesis Module and the NEBNext RNA Ultra Second Strand Synthesis Module (New England Biolabs, Ipswich, MA). Library preparation for an NGS platform using the TruSeq DNA Nano LT Library Prep Kit (Illumina, San Diego, CA) was performed according to the manufacturer's instructions. The NGS libraries were loaded onto an iSeq 100 i1 Reagent (Illumina) and sequenced on an iSeq 100 (Illumina). De novo assembly was performed using the CLC Genomics Workbench 12 (Qiagen, Hilden, Germany). BLASTN and BLASTX searches of the obtained contigs were conducted against INSD. Gaps in coverage were bridged by RT-PCRs with primer sets based on the contig sequences. The terminal sequences of the viral RNA segments were obtained by rapid amplification of the cDNA ends (RACE) as previously described (Li et al. 2005). Sequencing of the RACE and RT-PCR products was performed as described previously.
To detect orbiviruses that were first identified in Japan during the surveillance, oligonucleotide primers (Supplementary Table S1) were developed targeting the coding sequence of OC1 (outer capsid protein) for use in conventional RT-PCR assays. Before cDNA synthesis, the primer-template mix was heated at 98°C for 5 min followed by incubation on ice. A single-tube RT-PCR using the PrimeScript™ II High Fidelity One-Step RT-PCR Kit (Takara Bio, Shiga, Japan) was performed with the following parameters: cDNA synthesis at 45°C for 15 min, followed by heat inactivation at 94°C for 2 min, then 35 cycles of 98°C for 10 s, 55°C for 15 s, and 68°C for 10 s, and terminated at 4°C. The sequences of the PCR products were determined by Sanger sequencing.
The sequences obtained in this study are available in INSD under the accession numbers LC585872–585881, LC585908–585918, LC586079–586081, LC586145–586156, LC586727–586240, and LC586272–586276 (Supplementary Table S2). Phylogenetic analyses of the sequences aligned with MAFFT (Katoh et al. 2019) were performed using the maximum-likelihood method with 1000 bootstrap replicates in MEGA X (Kumar et al. 2018).
Results
Virus isolation and identification
A total of 308 bovine blood samples that were further separated to plasma and blood cells were used in virus isolation attempts (Table 1). The samples collected in 2014 tested negative for virus isolation using hamster cell lines (BHK-21 cells and HmLu-1 cells) in a previous study (Kato et al. 2016b). The blood sampling was conducted annually on Ishigaki, Kuroshima, Kohama, and Iriomote Islands during this study, but was conducted irregularly on Taketomi, Hateruma, and Yonaguni Islands (Table 1). No clinical signs were reported in the sentinel cattle at the sampling.
Eleven viruses were isolated from plasma, whereas 39 viruses were recovered from blood cells: in total, 50 isolates were obtained from the processed samples (Table 2 and Supplementary Table S2). In seven blood samples, same virus was isolated from both plasma and blood cells. However, each of these isolations was regarded as a single isolation event. Therefore, 43 blood samples tested positive for virus recovery. An orthobunyavirus, Peaton virus (PEAV; species Peaton orthobunyavirus, genus Orthobunyavirus, family Peribunyaviridae), was isolated, but the other viruses belonged to the genus Orbivirus of the family Reoviridae: two serotypes (12 and 16) of BTV, three serotypes (5, 6, and 7) of EHDV, two serotypes (DAGV and BCV) of PALV, Yunnan orbivirus (YUOV), Guangxi orbivirus (GXOV), and Yonaguni orbivirus (YONOV). Seventeen isolates including PEAV, BTV-16, EHDV-5, EHDV-6, EHDV-7, DAGV, and BCV were identified by previously reported conventional RT-PCRs that were virus/serotype specific (Supplementary Table S1). Six of the remaining isolates underwent NGS analysis and the obtained sequences underwent a homology search using the BLAST search program. The homology search results demonstrated that the NGS-analyzed samples included single isolates of BTV-12 and YUOV, two isolates of GXOV, and three isolates of a previously unknown virus. One of the unknown was recognized as the novel orbivirus (YONOV) by whole-genome analysis in a previous study (Murota et al. 2020). Sequences of the other two isolates were highly homologous with YONOV (data not shown). Based on the Seg3 sequences of YUOV, GXOV, and YONOV, virus-specific primer sets were designed and RT-PCRs with these primer sets sorted the 19 remaining unidentified viruses and found them to be 15 YUOV isolates, three GXOV isolates, and one YONOV isolate (Table 2 and Supplementary Table S2).
Arboviruses Recovered from Bovine Blood on the Yaeyama Islands Between 2014 and 2019
Number of positive cattle for each virus is given in parenthesis.
Most of the orbiviruses were isolated from washed blood cells with a few exceptions: four isolates of YUOV, two isolates of EHDV-7, and a single isolate of EHDV-5 from both plasma and blood cells; two isolates of BTV-16, a single isolate of EHDV-7, and a single isolate of YONOV from plasma only (Table 3 and Supplementary Table S2). Three isolates consisting of two DAGV and one BCV were obtained through the passage in the BHK-21 cell line, but the vast majority of orbiviruses were isolated through the passage in the mosquito cell line. Both the BHK-21 and C6/36 cell lines were sensitive to the recovery of DAGV. However, the recovery of PEAV and BCV was only successful through the BHK-21 cell line passage. No isolation of BTV, EHDV, YUOV, GXOV, or YONOV was observed through the passage in hamster cell lines. The HmLu-1 cell line was not sensitive to isolating arboviruses in this study.
Isolation Source and Sensitive Cell Lines of the Recovered Viruses
BCV, Bunyip creek virus; BTV, bluetongue virus; EHDV, epizootic hemorrhagic disease virus; DAGV, D'Aguilar virus; GXOV, Guangxi orbivirus; PEAV, Peaton virus; YONOV, Yonaguni orbivirus; YUOV, Yunnan orbivirus.
YUOV was most often recovered from bovine blood samples through surveillance; 16 isolations of the virus were achieved in four annual surveys. DAGV, GXOV, and YONOV were also recovered in three annual surveys. EHDV-5 was isolated in 2 consecutive years. YONOV, YUOV, GXOV, EHDV, and DAGV were isolated on multiple (3–4) islands. The recovery of PEAV, BTV-12, BTV-16, EHDV-6, EHDV-7, and BCV was less common, with each occurring in one location in an annual survey.
Sequence analysis and comparison of the obtained viruses
The partial Seg2 sequence of BTV-12 obtained in 2017 (ON-11/E/17 and ON-12/E/17) maintain 87.9–99.7% nt identity with that of available BTV-12 sequences (Supplementary Table S3). The sequences have a high identity with the homologous sequence of a recent Chinese BTV-12 isolate (G19-12-Llq) (99.7% nt identity and 100% aa identity) and a lower identity with that of a Japanese BTV-12 isolate (ON90-2) obtained in 1990 (95.9% nt and 95.6% aa identities). The phylogenetic analysis based on the partial sequence of BTV-12 Seg2 revealed that ON-11/E/17 and ON-12/E/17 are grouped with Australian, Indian, and Chinese BTV-12 isolates (Fig. 2). The partial Seg2 sequence of BTV-16 obtained in 2014 (ON-4/P/14 and ON-6/P/14) are identical. The complete coding sequence in Seg2 of ON-4/P/14 was determined by NGS analysis and has 73.5–98.8% nt identity with that of available BTV-16 sequences. Sequence comparison with other BTV-16 isolates showed a high identity with Japanese isolates obtained since 2001 (98.5–98.8% in nt and 98.6–98.9% in aa) (Supplementary Table S4). Phylogenetic analysis based on the BTV-16 Seg2 sequences showed that ON-4/P/14 forms a group with other Japanese BTV-16 isolates except older isolates in 1985 and 1990 (Fig. 3). The Japanese isolates are also closer to Indian BTV-16 isolates.

Phylogenetic analysis based on Seg2 of bluetongue virus serotype 12. Phylogenetic relationship of the OC1 coding sequences in Seg2 was inferred in MEGA X using the maximum-likelihood method and Kimura 2-parameter model. A discrete Gamma distribution was used to model evolutionary rate differences among sites. Node support value (%) were generated from 1000 bootstrap replicates. The scale bar indicates 0.02 substitution per site. Seg2 of ON-11/E/17 and ON-12/E/17 are depicted with black dots.

Phylogenetic analysis based on Seg2 of bluetongue virus serotype 16. Phylogenetic relationship of the OC1 coding sequences in Seg2 was inferred in MEGA X using the maximum-likelihood method and Tamura-Nei model. A discrete Gamma distribution was used to model evolutionary rate differences among sites. The rate variation model allowed for some sites to be evolutionarily invariable. Node support value (%) were generated from 1000 bootstrap replicates. The scale bar indicates 0.1 substitution per site. Seg2 of ON-4/P/14 is given in black dots.
The isolates of EHDV-5 obtained in 2 consecutive years, 2016 and 2017, (ON-11/E/16, ON-12/E/16, and ON-6/E/17) were almost identical in the partial sequence of Seg2 (99.8% nt identity). The sequence of ON-11/E/16 was most homologous with the corresponding sequence of the prototype EHDV-5 (CSIRO157) from Australia, which is the solo sequence of EHDV-5 Seg2 sequence available in INSD (90% nt and 94.8% aa identities). A partial Seg2 sequence of EHDV-6 obtained in 2015 (ON-3/E/14) has 89.3% nt and 94.7% aa identities with the corresponding sequence of the prototype EHDV-6 (CSIRO 753) from Australia (Supplementary Table S5). The sequence of ON-3/E/14 was identical with the homologous sequence of HG-1/E/15, which was detected from a diseased cow during an epizootic hemorrhagic disease outbreak in mainland Japan in 2015 (Kamomae et al. 2018). According to phylogenetic analysis based on Seg2 sequences, the Japanese EHDV-6 isolates are likely contained in a group that consists of Australian, Caribbean, and North American EHDV-6 isolates, although they form a separate clade (Fig. 4). The partial Seg2 sequence of EHDV-7 isolated in 2016 (ON-6/E/16) shared 72.1–96.9% nt identity with the homologous sequence of the EHDV-7 isolates previously characterized and has a high identity with the corresponding sequence of previous Japanese and Chinese EHDV-7 isolates (96.0–96.9% nt and 98.2–99.1% aa identities) (Supplementary Table S6). The Japanese and Chinese EHDV-7 isolates are phylogenetically closer to the Australian prototype strain (CSIRO 775) than Israeli EHDV-7 isolates (Fig. 5).

Phylogenetic analysis based on Seg2 of epizootic hemorrhagic disease virus serotype 6. Phylogenetic relationship of the OC1 coding sequences in Seg2 was inferred in MEGA X using the maximum-likelihood method and Tamura 3-parameter model. The rate variation model allowed for some sites to be evolutionarily invariable. Node support value (%) were generated from 1000 bootstrap replicates. The scale bar indicates 0.2 substitution per site. Seg2 of ON-3/E/14 is depicted with black dot.
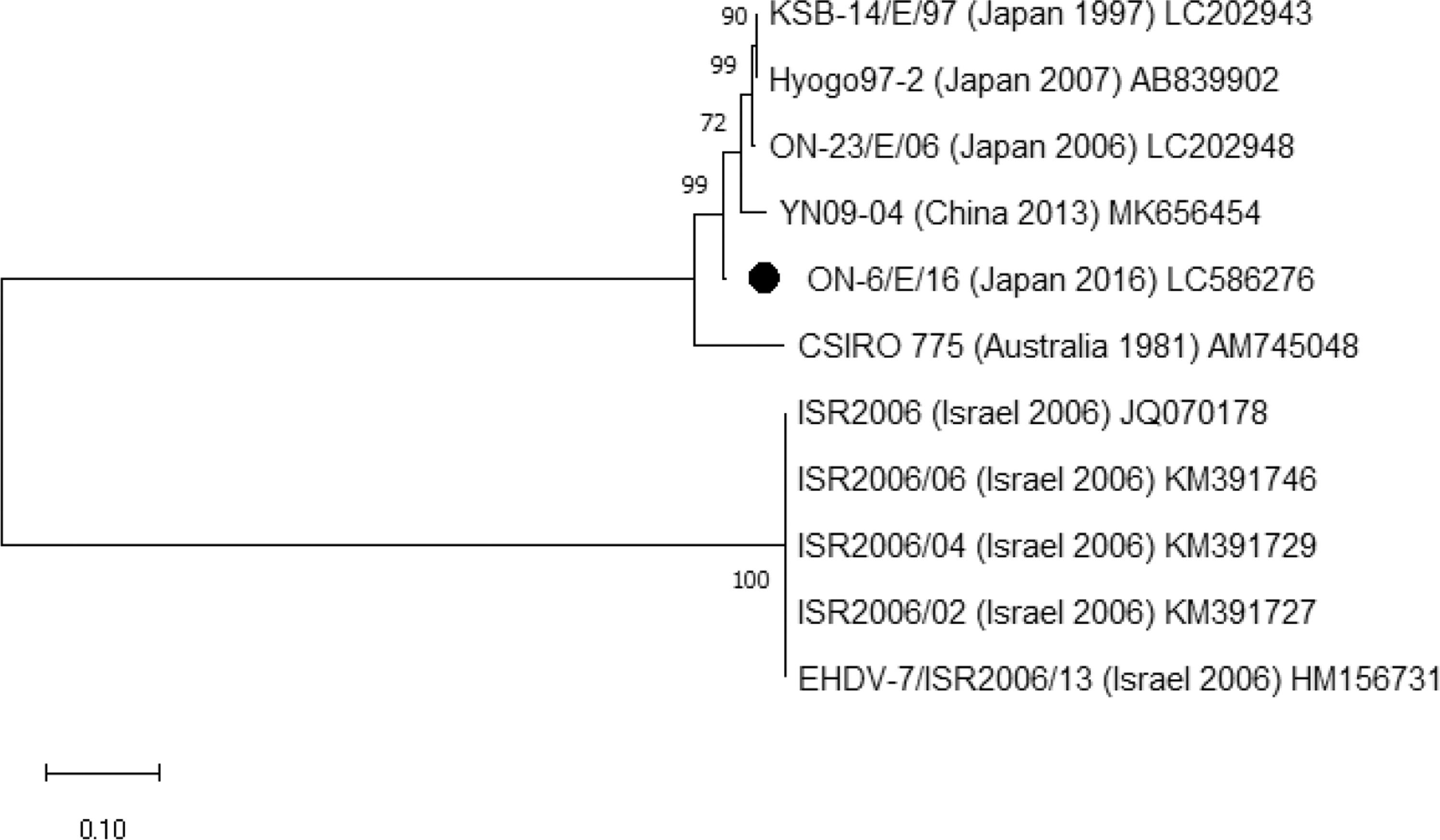
Phylogenetic analysis based on Seg2 of epizootic hemorrhagic disease virus serotype 7. Phylogenetic relationship of the partial Seg2 sequences was inferred in MEGA X using the maximum-likelihood method and Tamura 3-parameter model. The rate variation model allowed for some sites to be evolutionarily invariable. Node support value (%) were generated from 1000 bootstrap replicates. The scale bar indicates 0.1 substitution per site. Seg2 of ON-6/E/16 is given in black dots.
Of the three DAGV isolates, the 2017 and 2018 isolates (ON-3/E/17 and ON-5/E/18) were undistinguishable in the partial Seg2 sequences. However, the DAGV isolate obtained on Yonaguni Island in 2019 (ON-1/E/19) appeared to be less identical in the corresponding sequence with the aforementioned isolates (97.2% nt identity). These isolates were closely related with the Japanese DAGV isolates obtained in 1991, 2000, and 2001 (97.0–98.4% nt and 98.1–100% aa identities), but less related with other Japanese isolates obtained in 1987 and 2013 (91.9–93.4% nt and 97.6–98.1% aa identities) and the prototype DAGV (B8112) from Australia (90.4–90.8% nt and 95.8–96.2% aa identities) (Supplementary Table S7). Phylogenetic analysis of the partial Seg2 sequence of DAGV isolates revealed that Japanese DAGV isolates are roughly divided into two groups and the isolates characterized in this study are included in a group represented from ON91-5 (Supplementary Fig. S1). Sequence comparison of the partial Seg2 sequence between BCV obtained in 2017 (ON-14/E/17) and other BCV isolates showed a high identity with previous Japanese isolates (98.9–99.3% nt and 99.9–100% aa identities) and a lower identity with the prototype BCV (CSIRO58) from Australia (87.5% nt and 94.4% aa identities). Phylogenetic analysis also revealed the close relationship of ON-14/E/17 with the other Japanese BCV isolates (Supplementary Fig. S2).
The whole-genome sequences of one isolate each of YUOV and GXOV obtained in this study were determined. A YUOV isolate, ON-4/P/18, which was isolated from bovine blood cells collected on Ishigaki Island in 2018, was genetically similar to the prototype of YUOV (YOV-77-2) (Table 4). Each segment shares 92.3–98.6% nt and 95.0–100% aa identities between the two isolates. Seg3, Seg5, and Seg7, which encode OC1, TuP (nonstructural protein forming tubes), and ViP (viral inclusion body matrix protein), respectively, showed relatively variable (92.3–93.8% nt) identities between the isolates. In comparison with Indonesian YUOV isolates obtained in 1981, higher variations were observed in Pol (RNA-dependent-RNA-polymerase), OC1, and VRP (virus release protein) (74.2–87.2% nt and 86.2–94.2% aa identities). The partial sequence (446 nt) of Seg3 was obtained from other YUOV isolates derived on Ishigaki and Iriomote Islands in 2015, on Ishigaki and Yonaguni Islands in 2016, and on Ishigaki Island in 2019. The sequences were identical with the corresponding region of ON-4/P/18.
Comparison Between Yunnan Orbivirus ON-4/P/18 and YOV-77-2
The whole-genome sequence of ON-2/E/14 of GXOV, which was isolated from bovine blood cells collected on Kuroshima Island in 2014, showed high identities (97.2–99.6% nt and 99.2–100% aa in each segment) with the published sequence of GXOV V172_GX_2015 (Table 5). Comparison of the partial sequence (1147 nt) of Seg3 obtained from three other GXOV isolates derived on Iriomote Island in 2014 and on Yonaguni Island in 2015 and 2018 indicated the presence of a small variation in the corresponding sequence of ON-2/E/14 (99.3–99.5% nt identity).
Comparison Between Guangxi Orbivirus ON-2/E/14 and V172/GX/2015
In comparison with the 2015 isolate of YONOV, ON-7/E/15, other isolates derived on Kohama and Iriomote in 2017 and on Ishigaki in 2018 had several mutations (1 or 7 nt) in the partial Seg3 sequence (794 nt). Although retrievable sequences are limited, phylogenetic analyses of three mosquito-borne orbiviruses revealed that the domestic isolates of each virus are less variable (Supplementary Figs. S3, S4, S5).
Discussion
This study demonstrated that a variety of orbiviruses circulated on the Yaeyama Islands between 2014 and 2019. Unlike the previous surveillance (Kato et al. 2016b), the mosquito cell line C6/36 was added to our surveillance to increase the efficacy of the virus isolation. Forty-two orbivirus isolates were recovered through the passage of the blood samples in C6/36 cells, and the obtained viruses also contained three potential mosquito-borne orbiviruses and two serotypes of EHDV, all of which were new in Japan. As reported from previous surveillance efforts in other regions (Gard et al. 1988b, Wang et al. 2011, Cooper et al. 2014, Yang et al. 2018), the mosquito cell line covered a wider range of orbiviruses in the attempt at virus isolation. In contrast, orthobunyaviruses cannot induce obvious CPEs in the infected C6/36 cells despite their continuous and efficient replication (Han 1981, Elliott and Wilkie 1986). Mammalian and mosquito cell lines could be complementary to each other in the field of arbovirus surveillance.
Three serotypes (5, 6, and 7) of EHDV were obtained in this surveillance effort. The Seg2 sequence of EHDV-6 isolated on Ishigaki Island in 2014 was identical with that of EHDV-6 detected in cattle with hemorrhagic disease in the central part of mainland Japan in 2015. Although no epizootic hemorrhagic disease in Yaeyaman cattle was reported, it was confirmed that the virus was prevalent on the Yaeyama Islands before its incursion to mainland Japan. EHDV-5 was also isolated from sentinel cattle in Australia and mainland China (Gard et al. 1988b, Yang et al. 2020), and its seroprevalence in Indonesian domestic ruminants was demonstrated (Sendow et al. 1991). Its relationship to clinical signs in ruminants has not been determined so far (Savini et al. 2011) as shown in this study. EHDV-7 has been repeatedly detected in Japan and mainland China so far (Shirafuji et al. 2017, Qi et al. 2019) and its association with stillbirth and abortion in cattle was suggested (Ohashi et al. 1999).
Other Culicoides-borne orbivirus species, PALV and BTV, were obtained during the study. As reported in the previous surveillance (Kato et al. 2016b), two serotypes of PALV, DAGV and BCV, were isolated from cattle on the Yaeyama Islands. The findings indicated that these viruses have been continuously prevalent in the area surrounding the islands. It should be noted that DAGV is thought to be involved in congenital abnormalities in calves (Ohashi et al. 2004a). However, BCV has not been recognized as an etiological agent in diseased cattle. Both the BTV-12 and BTV-16 that were obtained in this study were previously isolated in the southwestern islands of Japan in 1990 (Shirafuji et al. 2012). Isolations and seroprevalence of these serotypes from domestic ruminants have also been reported in other regions in East Asia (Zhang et al. 1999, Lee et al. 2010, Hwang et al. 2019). These serotypes probably have been circulating in East Asia and/or its neighboring regions and have occasionally been introduced to the Yaeyama Islands.
During the surveillance, three orbiviruses, YUOV, GXOV, and YONOV, which are phylogenetically clustered with mosquito-borne orbiviruses, were first isolated in Japan. The NGS platform was of great use in identifying these viruses because their prevalence had been unexpected in Japan and no available method of PCR-based identification was prepared for them. Only the C6/36 cell line was sensitive for these orbiviruses in the virus isolation attempt. Several orbiviruses grouped as mosquito-borne orbiviruses were initially isolated by passages through mosquito cell lines and could not replicate well in mammalian cell lines (Attoui et al. 2005, 2009, Ahasan et al. 2019). Owing to the utilization of C6/36 cells in the attempts to isolate viruses since 2014, the circulation of YUOV, GXOV, and YONOV could have been detected in the Yaeyama Islands.
The first isolation of YUOV was recorded in mainland China in 1997 (Attoui et al. 2005). YUOV has also been found in Peru and Indonesia (Attoui et al. 2009, Sadeghi et al. 2017). The virus was recovered from blood and/or brains of diseased donkey, cattle, sheep, and dogs with neurological signs in addition to field-collected mosquitoes in Peru (Attoui et al. 2009). The whole-genome sequence of ON-4/P/18 demonstrated its close genetic relatedness with the Chinese isolate. The other orbiviruses, GXOV and YONOV, were first isolated from cattle in southern China and Yonaguni Island, respectively, in 2015 (Yang et al. 2018, Murota et al. 2020). GXOV was proposed to be assigned to a novel orbivirus species based on its lower genetic identity with known orbivirus species. In contrast, YONOV was considered a novel serotype of Mobuck virus that is distributed in North America (Cooper et al. 2014). The GXOV isolate obtained in this study has high identities with the Chinese isolate through all segments, suggesting that GXOV originated from the same genetic pool and rapidly spread in the southern part of East Asia. At present, the potential involvement of GXOV and YONOV in animal diseases could not be excluded because of their genetic relationship with the pathogenic orbiviruses such as YUOV and Peruvian horse sickness virus suspected to cause neuronal disorders (Attoui et al. 2009).
Although we performed blood collection once per year during the surveillance, many orbiviruses were isolated from bovine blood samples. Long-lasting viremia of BTV, EHDV, and PALV in cattle has been reported (Gard et al. 1988b, Singer et al. 2001, Savini et al. 2011). In addition, Middle Point virus, which is a serotype of YUOV, persistently infected cattle for up to 5 months (Cowled et al. 2007). Moreover, the mild climate in the islands, which contributes to the maintenance of vector activities all through the year, might be beneficial for maintaining the transmission cycle of orbiviruses.
Culicoides oxystoma, which appeared to be a competent vector of EHDV, BTV, DAGV, and BCV (Yanase et al. 2005, Prasad et al. 2009, Kato et al. 2016a), was distributed on the Yaeyama Islands (Wada et al. 1996). Other potential BTV vectors, such as Culicoides wadai, Culicoides actoni, and Culicoides asiana (Daniels et al. 2009, Yanase et al. 2010, Gopurenko et al. 2015), were also recorded in the islands (Wada et al. 1996). YUOV has been isolated from Culex tritaeniorhynchus in China (Attoui et al. 2005, Wang et al. 2011). However, the virus was recovered from Aedes mosquitoes in Peru (Méndez-López et al. 2015), suggesting that more mosquito species belonging to multiple genera have the vector capacity for YUOV. Although there has been no information about the vector species of GXOV and YONOV, the phylogenetic analysis indicated that these viruses are transmitted by mosquitoes (Yang et al. 2018, Murota et al. 2020). A rich fauna of mosquitoes on the Yaeyama Islands was reported (Taira et al. 2012) and some of the indigenous species preferred to feed on cattle (Tamashiro et al. 2011).
Previous vector studies demonstrated that Culicoides biting midges and mosquitoes were active much of the year on the islands (Hoshino 1985, Toma et al. 2002, Higa et al. 2007). Together with the long-lasting viremia, the unique ecosystem of the islands might have contributed to the maintenance of infection cycles of orbiviruses in consecutive years. As it is, the genetic variation of each YUOV, GXOV, and YONOV isolated during the study period was quite small, suggesting their establishment on the Yaeyama Islands. In addition, DAGV and EHDV-5 were isolated on the islands in 2 consecutive years.
It is of interest that the Yaeyama Islands are at a remote distance from other regions and their land area is small, although the circulation of Culicoides-borne and mosquito-borne viruses has often been reported (Aizawa et al. 2008, Hayama et al. 2016, Kato et al. 2016b, 2018). Although livestock animal movement between Japan and neighboring countries has been prohibited under the governmental quarantine system, the genetic analysis of the obtained viruses suggested an epidemiological relationship with these countries.
Previous molecular epidemiological studies of bovine ephemeral fever virus and backward trajectory analyses of an outbreak of bovine ephemeral fever on the Yaeyama Islands indicated that the viruses come from Taiwan and/or Southeast Asia (Kato et al. 2009, Niwa et al. 2015, Hayama et al. 2016). The phylogenetic analysis in this study also suggested the epidemiological relationships between the Yaeyama Islands and its neighboring regions, despite limited genomic sequence data of Asian isolates being available. Historically, the long-distance dispersal of Culicoides biting midges and mosquitoes, and its contribution to arbovirus spreading have been indicated since the mid-twentieth century (Elbers et al. 2015). These findings suggested that many arboviruses might have been introduced to the Yaeyama Islands through infected vectors that were brought by the air stream from overseas. It may have reflected the diversity of arboviruses in the Yaeyama Islands.
Conclusions
This study demonstrated the current status of Culicoides- and mosquito-borne orbiviruses on the Yaeyama Islands and suggested a rich fauna of these viruses in neighboring regions, such as other subtropical islands in Japan, Taiwan, Southern China, and Southeast Asia. In comparison with Culicoides-borne orbiviruses, the clinical and consequent economic significance of mosquito-borne orbiviruses remains uncertain. This knowledge gap should be filled by further etiological research.
Footnotes
Acknowledgments
We express our sincere thanks to the staff of Yaeyama Livestock Hygiene Service Center for their support. We also wish to acknowledge Tomoko Kato for her technical assistance in attempts at virus isolation.
Authors' Contribution
K.M.: investigation, data curation, writing—original draft; K.I.: investigation, resources; Y.M.: investigation, resources; M.A.: investigation, resources; Y.S.: investigation, writing—review and editing; H.S.: investigation, writing—review and editing; D.K.: investigation, writing—review and editing; H.I.: methodology, writing—review and editing; T.Y.: conceptualization, investigation, data curation, writing—original draft, writing—review and editing, supervision.
Ethics Statement
No human subjects were used in this study. Bovine blood sampling was conducted as a part of the epidemiological investigation based on the domestic animal infectious disease control law in Japan, and permission to use the samples was granted by the animal health authority of Okinawa Prefecture. This study was performed under guidelines provided by the National Institute of Animal Health, NARO, and did not include any animal experiments. Therefore, further approval by an ethical committee was not required.
Author Disclosure Statement
No conflicting financial interests exist.
Funding Information
This study was conducted as part of a research project on “Development of Management Technologies for the Risk of Introduction of Livestock Infectious Diseases and Their Wildlife-borne Spreads in Japan” (JP J008617. 18065101) funded by the Ministry of Agriculture, Forestry, and Fisheries of Japan.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
Supplementary Table S7
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.