
Abstract
Introduction:
We screened host-collected ticks for tick-borne viruses, including those recently documented as human pathogens.
Methods:
During 2020–2021, ticks removed form cattle, sheep, dogs, and cats in 11 provinces in 5 geographically distinct regions of Anatolia were identified, pooled, and screened using pan-nairovirus, pan-flavivirus and individual assays for Jingmen tick virus (JMTV), and Tacheng tick virus 1 and 2 (TcTV-1 and TcTV-2).
Results:
A total of 901 tick specimens, comprising 6 species were included. Rhipicephalus sanguineus complex was the most abundant species (44.1%), followed by Rhipicephalus bursa (38.3%), Haemaphysalis parva (7.2%), and others. The specimens were screened in 158 pools with 12 pools (7.6%) being positive. Crimean–Congo hemorrhagic fever virus (CCHFV) lineage Europe 2 (genotype VI) sequences were detected in R. bursa in five (3.2%) of the pools, with similar prevalences in central and Mediterranean Anatolian provinces. JMTV was identified in four R. bursa and one Rhipicephalus turanicus pools, collected from Mediterranean and southeastern Anatolia, with a CCHFV and JMTV coinfected R. bursa pool. The JMTV segment 1 sequences formed a separate cluster with those from Turkey and the Balkan peninsula in the maximum likelihood analysis. TcTV-2 was detected in two Dermacentor marginatus specimens (1.3%) collected in central Anatolia, with nucleocapsid sequences forming a phylogenetically segregated group among viruses from humans and ticks from China and Kazakhstan.
Discussion:
CCHFV Europe 2 was initially documented in ticks from central Anatolian locations, where related orthonairoviruses had been previously recorded. Ongoing activity and a wider distribution of JMTV and TcTV-2 were observed. These viruses should be screened as potential etiological agents in human infections associated with tick bites.
Introduction
Ticks act as biological vectors of several viral pathogens, causing human and livestock diseases with significant public health and economic impact (Mansfield et al. 2017). Tick-borne infections are frequently of zoonotic origin, where the causative agent is maintained in natural cycles involving vector ticks and animal hosts, with humans rarely contributing to the circulation (Kazimirova et al. 2017). The tick feeding behavior, opportunities for vertebrate contact, as well as climatic and other ecological factors contribute to the complex interplay affecting virus dispersion and disease emergence or resurgence.
Viruses transmitted to vertebrates by ticks are heterogeneous with diverse characteristics (Mansfield et al. 2017). Tick-borne viruses with RNA genomes include the flaviviruses, tick-borne encephalitis virus (TBEV), Omsk hemorrhagic fever virus, Kyasanur forest disease virus, Powassan virus, and Alkhurma hemorrhagic fever virus; the orthonairoviruses Crimean–Congo hemorrhagic fever virus (CCHFV) and Nairobi sheep disease virus. The infections caused by severe fever with thrombocytopenia syndrome virus and Heartland virus (Nairoviridae: Bandavirus) have been described since 2011, becoming the first well-characterized tick-borne human viral pathogens following a gap of decades ( Zhan et al. 2017, Brault et al. 2018).
In addition, flaviviruses with segmented genomes, such as Jingmen tick virus (JMTV) and Alongshan virus, as well as Tacheng tick virus 1 (TcTV-1; Nairoviridae: Orthonairovirus) and 2 (TcTV-2; Phenuiviridae: Uukuvirus) have recently been reported to cause human infections (Jia et al. 2019, Wang et al. 2019, Liu et al. 2020, Dong et al. 2021).
Epidemiological changes for the known agents and emergence of novel tick-borne viruses are estimated to occur, facilitated by anthroponotic and climatic factors acting on the global scale (Vayssier-Taussat et al. 2015). Given the paucity of specific therapeutics and vaccines, surveillance and pathogen screening in vectors are of paramount importance for timely identification of pathogen activity within a given region and implementation of control measures in tick-borne virus infections.
Located in the Palearctic zone, Turkey is a Mediterranean country on the lands of the Anatolian peninsula. It serves as a natural hub for the dissemination of vector-borne viruses among old world continents, due to its close association with bird migration routes. Diverse ecological conditions supporting a rich fauna of vectors and amplification hosts are recorded in distinct geographic regions of Anatolia. CCHFV is the eminent endemic tick-borne infectious agent in Turkey, with annual cases ranging between 343 and 1318 during 2008–2017 (Ergünay et al. 2020b).
Aside from scarce data on TBEV, screening and metagenome-based investigations have identified several viruses in ticks, including JMTV and TcTV-2, as well several known and novel flavi-like, chu, nairo, and phenuiviruses (Brinkmann et al. 2018, Dinçer et al. 2019). This study was carried out to screen for pathogenic tick-borne viruses previously documented or likely to circulate in Anatolia, specifically focusing on those recently described as human pathogens.
Materials and Methods
Tick collection and processing
Specimen collections were carried out at locations in Çankırı, Kayseri, and Konya provinces in central Anatolia, İzmir, and Kutahya in the Aegean region; Antalya and Burdur in the Mediterranean region; Malatya and Bingol in eastern; and Adıyaman and Şanlıurfa in southeastern Anatolia, during 2020–2021. Adult ticks were removed from infested animals comprising cattle (Bos taurus), sheep (Ovis aries), dogs (Canis familiaris), and cats (Felis catus domesticus) during routine visits and vaccinations at farms, clinics, or animal shelters, with informed consent and cooperation of the owners or caretakers without further intervention. Local Ethics Committee approval was not required in this setting.
Collected ticks were transferred in dry ice and morphologically identified to genus or species level, using appropriate taxonomic keys (Filippova 1997, Walker et al. 2000, 2003, Apanaskevich and Horak 2008, Estrada-Pena et al. 2004).
Subsequently, they were pooled up to 1–15 individuals according to collection site and species and developmental stage, to be stored at −80°C. The pools were ground by vortexing with tungsten carbide or stainless steel beads (QIAgen, Hilden, Germany), in 500–700 μL of Eagle's Minimum Essential Medium, supplemented with 5% fetal bovine serum. Following a 4-min centrifugation at 4000 rpm, nucleic acids were purified from the supernatant using the High Pure Viral Nucleic Acid Kit (Roche Diagnostics, Mannheim, Germany), with subsequent complementary DNA (cDNA) synthesis with random hexamers, using the RevertAid First-Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Hennigsdorf, Germany), as recommended by the manufacturers.
Virus screening
A selection of PCR-based assays targeting all major groups of pathogenic tick-borne viruses were employed for screening (Table 1). For orthonairoviruses, a single-round PCR with primers targeting RNA-dependent RNA polymerase (RdRp) central motif A on the genome segment L, was used (Honig et al. 2004). This assay generates amplicons from CCHFV and Nairobi sheep disease virus, as well as Qalyub, Bandia, Raza, Farallon, Punta Salinas, Abu Hammad, Abu Mina, Hazara, Dugbe, Erve, and Tillamook viruses.
PCR Assays Used for Screening
Wobble bases in oligonucleotides are: I:Inosine; M:A/C; N:A/T/G/C; R:A/G; Y:C/T; W:G/C. Cycling conditions were described in the original references.
Primer positions and PCR product size vary according to the particular virus isolate for each generic PCR.
According to JMTV isolate SY84 (NC024113), Tacheng Tick Virus 1 strain TC253 (NC031286), and Tacheng Tick Virus 2 strain TC252 (NC055426).
JMTV, Jingmen tick virus; RdRp, RNA-dependent RNA polymerase.
Flavivirus screening in ticks was carried out through another pan-virus assay incorporating degenerated primers to amplify the NS5 conserved region (Vazquez et al. 2012). This assay amplifies all major tick and mosquito-borne pathogenic flaviviruses, including TBEV, West Nile virus (WNV), Dengue virus, Yellow fever virus, Murray Valley encephalitis virus, Saint Louis encephalitis virus, and Usutu virus, as well as mosquito-specific strains, with a detection limit of 40 median tissue culture infective dose (TCID50) per reaction.
These assays have been instrumental in detecting CCHFV, Tamdy virus, WNV, mosquito-associated flaviviruses, and several novel viruses described in Anatolia ( Öncü et al. 2018, Akıner et al. 2019, Ergünay et al. 2020a). Finally, recently described tick-borne viruses, JMTV, TcTV-1, and TcTV-2 were screened individually by nested assays targeting NS5-like or nucleocapsid protein on the segment 1 and segment S, respectively (Table 1). CCHFV strain Ank-2 (GenBank acc, no. MK309333), TBEV strain Hypr, or previously processed tick pools (Brinkmann et al. 2018, Dinçer et al. 2019) were used as positive controls. Amplified products were subjected to electrophoresis in 1.2 − 1.7% agarose gels and visualized in a ChemiDoc XRS+imaging system (Bio-Rad Laboratories, Munich, Germany).
Sequencing and phylogenetic analysis
Products of expected size in each assay were cleaned up using the PureLink PCR Purification Kit (Thermo Fisher Scientific) for sequencing using an ABI PRISM 3500xL Dx Genetic Analyzer (Thermo Fisher Scientific) and the BigDye Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific). Geneious (version 11.1.5; Biomatters Ltd., Auckland, New Zealand) was used for handling raw sequence data. Similarity searches in the National Center for Biotechnology Information (NCBI) databases were carried out by BLASTn and BLASTn optimized for highly similar sequence (MEGABLAST) algorithms (Altschul et al. 1990).
Nucleotide and deduced amino acid sequence alignments and pairwise comparisons were generated using CLUSTAL W (Thompson et al. 1994). MEGAX was used for estimating optimal substitution model on individual alignments and to infer evolutionary history according to the Bayesian information criterion (Kumar et al. 2018).
Results
Tick collection and processing
We collected a total of 901 tick specimens, comprising 6 species (Table 2). The specimens comprised 272 ticks (30.2%) from southeastern Anatolian, 222 ticks (24.6%) from Mediterranean, 202 ticks (22.4%) from central Anatolian, 167 ticks (18.5%) from Aegean, and 38 ticks (4.2%) from eastern Anatolian provinces. The most abundant tick species was Rhipicephalus sanguineus complex (n = 397; 44.1%), followed by Rhipicephalus bursa (n = 345; 38.3%), Haemaphysalis parva (n = 65; 7.2%), and others (10.4%) (Table 2). The pools, 158 in total, originated from central Anatolian (37, 23.4%), Aegean (46, 29.1%), Mediterranean (30, 18.9%), eastern Anatolian (10, 6.3%), and south-eastern Anatolian provinces (35, 22.2%).
Distribution of the Specimens According to Species and Collection Sites
Virus detection
A total of 12 pools (12/158, 7.6%) were positive in virus screening. Positive pools originated from Antalya (n = 7, 58.3%), Konya (n = 2, 16.7%), Çankırı (n = 2, 16.7%), and Şanlıurfa (n = 1, 8.3%) provinces (Table 3). The pools comprised R. bursa (n = 9; 75%), Dermacentor marginatus (n = 2; 16.7%), and Rhipicephalus turanicus (n = 1; 8.3%) specimens collected from O. aries (n = 9; 75%) and B. taurus (n = 3; 25%). Reactive screening assays included JMTV (n = 5), generic nairovirus (n = 5) and TcTV-2 (n = 2). A single pool of R. bursa specimens collected in Antalya province (pool 58) was positive for JMTV as well as generic nairovirus assay. Generic flavivirus and TcTV-1 assays remained negative in all specimens.
Tick Pools with Detectable Viruses
CCHFV, Crimean–Congo hemorrhagic fever virus; TcTV-2, Tacheng tick virus 2.
Sequencing of the amplified products revealed CCHFV in all generic nairovirus reactive pools and confirmed virus identity in JMTV and TcTV-2 reactive pools. Virus detection rates according to tick species and collection region are provided in Table 4. The CCHFV sequences were detected in pools of female R. bursa specimens from Konya (central Anatolia) and Antalya (Mediterranean Anatolia) provinces (Table 3).
Virus Detection Rates According to Tick Species and Collection Region
Expressed as positive/total pools (percentage).
Describes positivity for the particular tick species collected from the region.
Pairwise comparisons (464 bp) revealed an intramural diversity of 1.8–3.2% and 95.2–97.7% identities to CCHFV lineage Europe 2 (AP92) viruses, previously detected in Anatolia. The deduced amino acid sequence of the partially amplified CCHFV RdRp was identical, except for the I2353V variation observed in pool 35. Phylogeny reconstruction revealed grouping of the sequences with the CCHFV Europe 2 (AP92) lineage viruses from Greece, Bulgaria, and Turkey, distinct from Europe 1, Asia, or Africa-oriented lineages (Fig. 1).
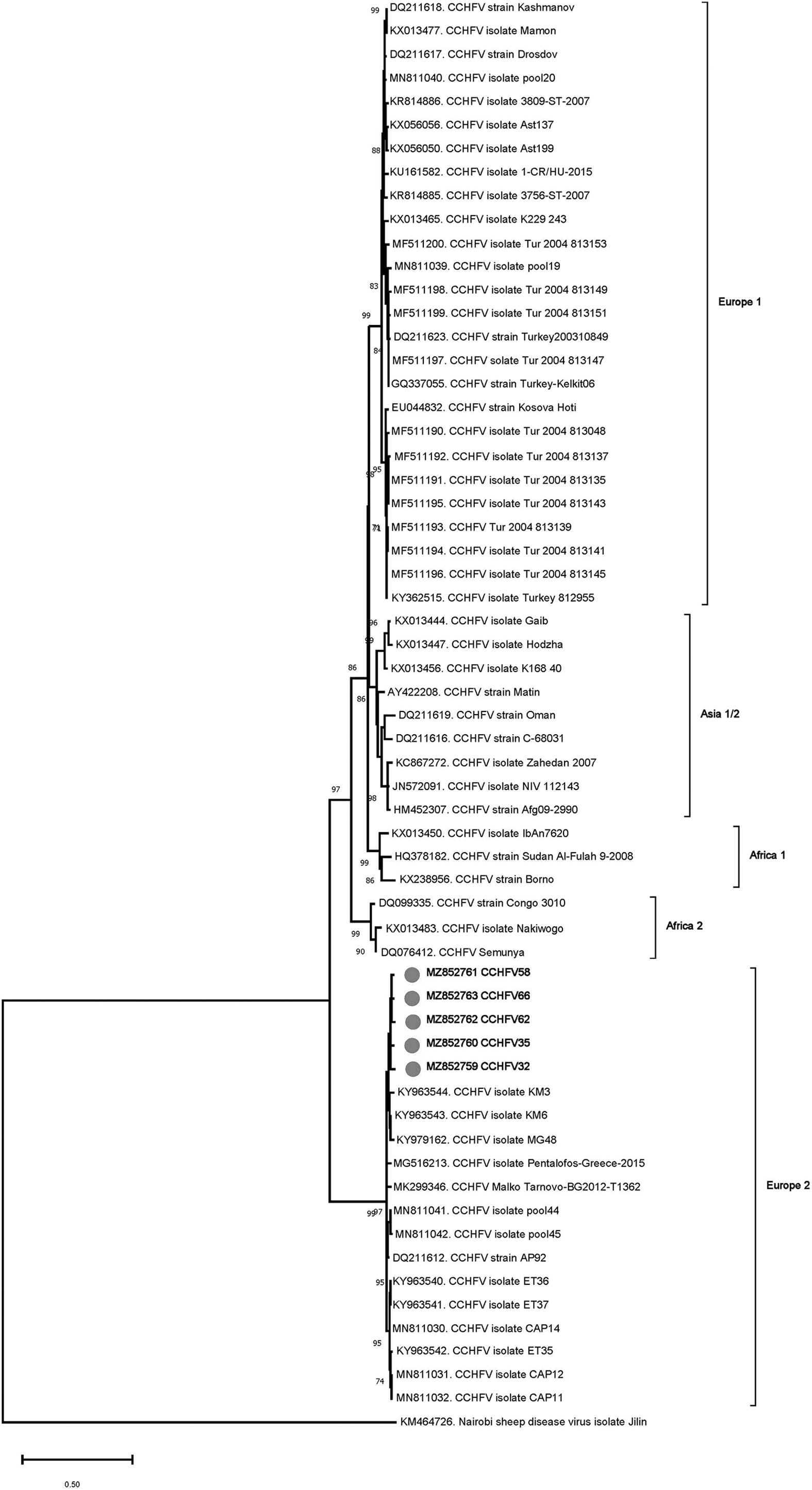
Maximum likelihood analysis of the CCHFV L segment sequences (445 nucleotides). The tree is constructed using GTR model, gamma distributed with invariant sites (G+I) for 500 replications. Viruses are indicated by GenBank accession number, abbreviation, isolate/strain identifier, and main lineage. The sequences characterized in the study are marked and in bold. Bootstrap values higher than 60 are provided. Nairobi sheep disease virus serves as an outgroup. CCHFV, Crimean–Congo hemorrhagic fever virus; GTR, General Time Reversible.
We further detected JMTV segment 1 sequences (368–403 bp) in 5 pools of female R. bursa and male R. turanicus ticks, collected from sheep in Antalya (Mediterranean Anatolia) and from bovine in Şanlıurfa (southeastern Anatolia), respectively. The sequences were also highly similar, with a maximum of 0.6% diversity. The sequences obtained from pools 50, 64, and 67 were identical. Pairwise comparisons showed 89.6–95.9% nucleotide identities with the JMTV isolates previously characterized in Turkey. The deduced amino acid sequence of the partial NS5-like protein was identical, with all sequences carrying N490 and A606 variations, previously reported from Turkey and other regions. In the maximum likelihood tree, the sequences remained distinct and formed a separate cluster with those from Turkey and the Balkan peninsula (Fig. 2).
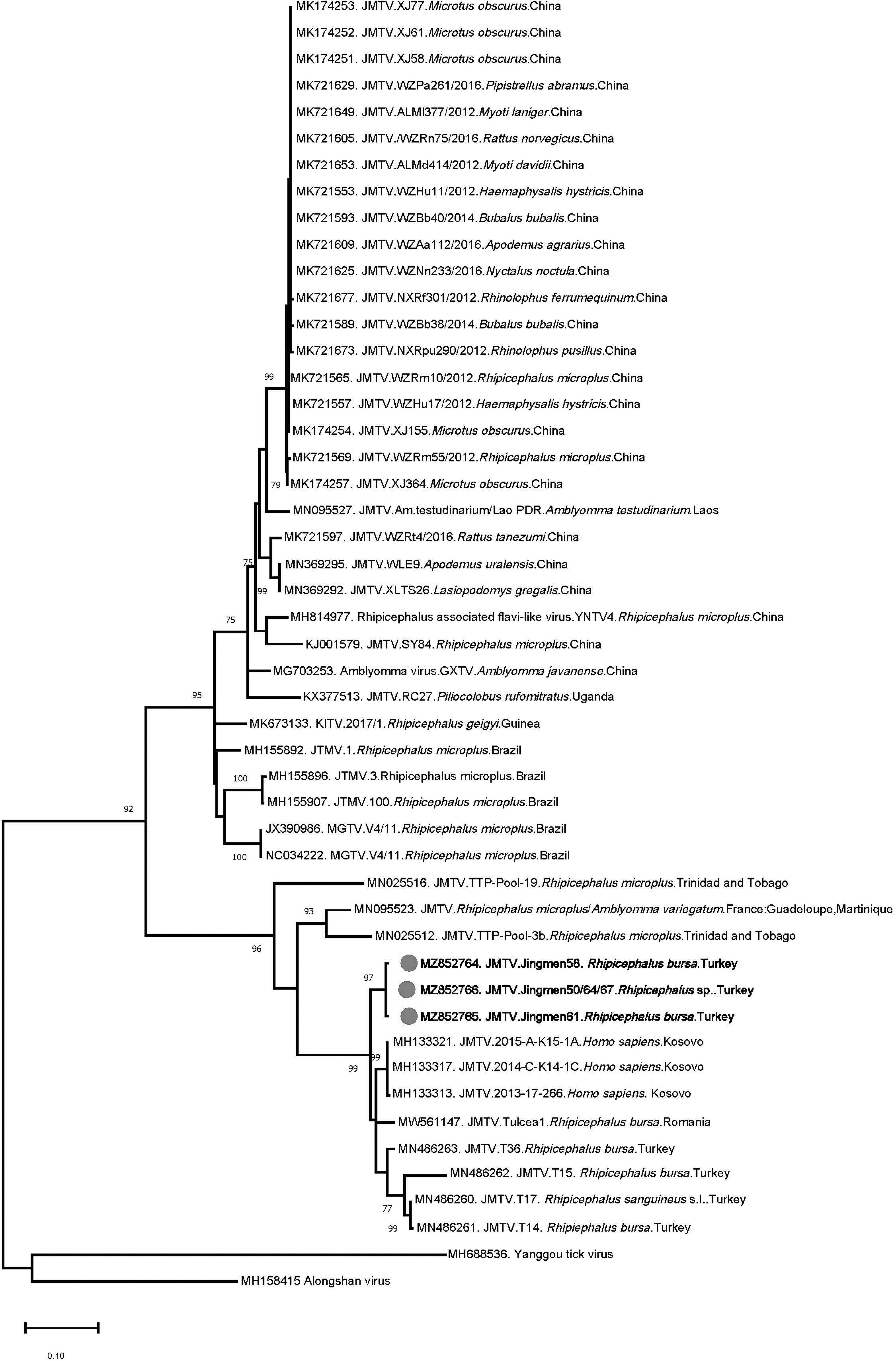
Maximum likelihood analysis of the JMTV segment 1 sequences (368 nucleotides). The tree is constructed using Tamura-Nei model for 500 replications. Viruses are indicated by GenBank accession number, abbreviation, isolate/strain identifier, tick species, and country of origin. The sequences characterized in this study are marked and in bold. Bootstrap values higher than 60 are provided. Yanggou tick virus and Alongshan virus are included as outgroups. JMTV, Jingmen tick virus.
Finally, TcTV-2 sequences were detected from Çankırı province (central Anatolia), in single female and male D. marginatus specimens collected and processed individually. Revealing 0.5% nucleotide diversity and identical deduced amino acid sequence, 93.4–97.6 identities with all globally available TcTV-2 strains were observed in pairwise comparisons. The sequences formed a well-defined phylogenetically segregated group among viruses detected in ticks and infected individuals from China and Kazakhstan (Fig. 3).
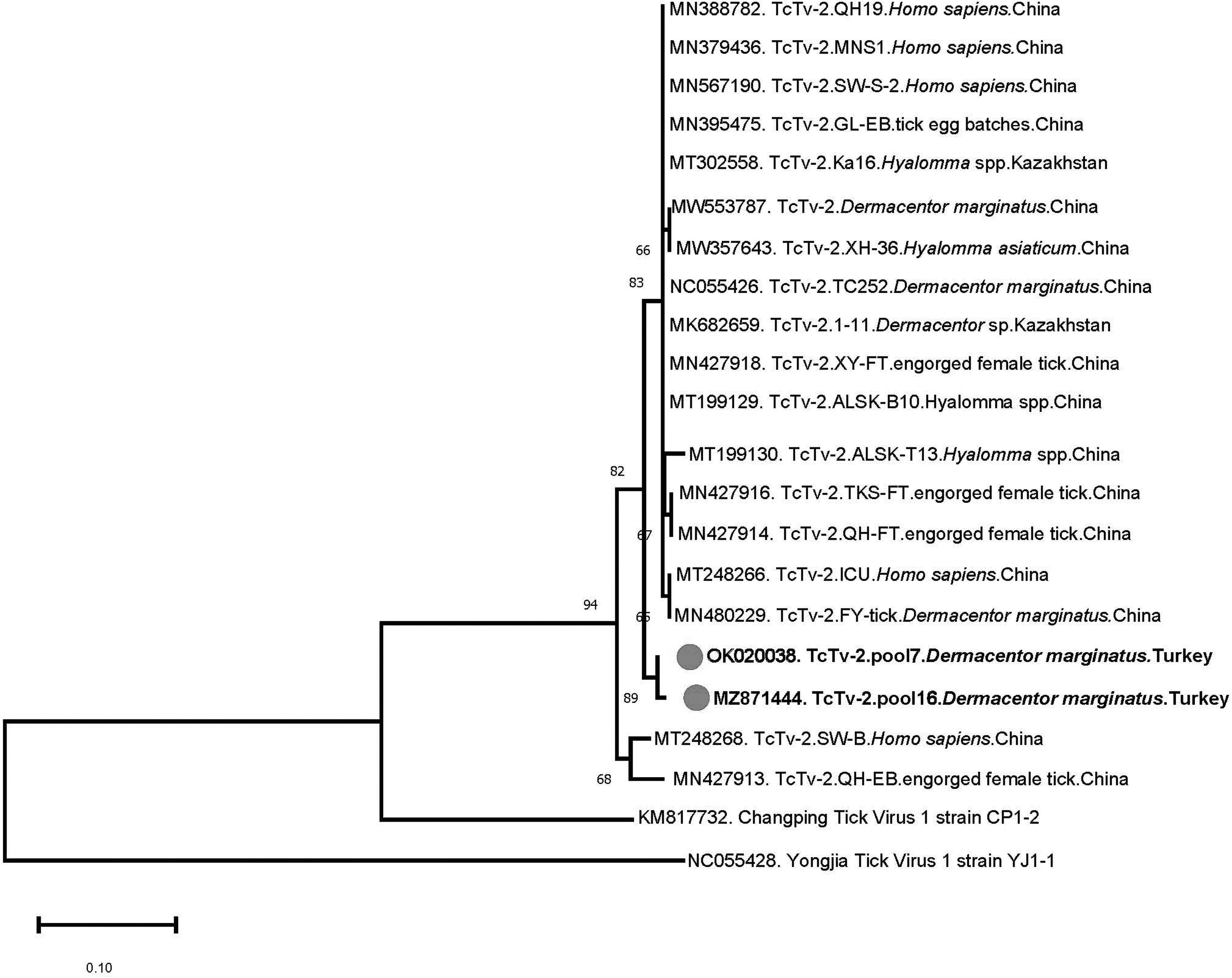
Maximum likelihood analysis of the TcTV-2 S segment sequences (227 nucleotides). The tree is constructed using Tamura 3-parameter model for 500 replications. Viruses are indicated by GenBank accession number, abbreviation, isolate/strain identifier, host/tick species, and country of origin. The sequences characterized in this study are marked and in bold. Bootstrap values higher than 60 are provided. Yongjia tick virus serves as outgroup. TcTV-2, Tacheng tick virus 2.
Discussion
In this study, we aimed to screen for tick-borne viruses, recently described to cause human infections as well as endemic and previously documented pathogenic viruses, in specimens from diverse regions in Anatolia. It also serves as a follow-up to our metagenomic investigations in ticks, revealing a wide spectrum of viruses, including newly described pathogens (Brinkmann et al. 2018, Dinçer et al. 2019). The tick collection comprised 901 specimens belonging to 6 species, obtained from domesticated animals from 11 provinces in 5 geographically distinct regions of Anatolia (Table 2). Pan-virus assays were employed for screening orthonairo and flaviviruses, along with individual nested PCRs for JMTV, TcTV-1, and TcTV-2. Virus detection was accomplished in 12 pools (7.6%) and product sequencing revealed CCHFV, JMTV, and TcTV-2 (Table 3).
We detected CCHFV lineage Europe 2 (genotype VI) sequences exclusively in R. bursa pools in 3.2% of the specimens, with similar prevalences observed in central and Mediterranean Anatolia (Table 4). The sequences were relatively conserved and grouped with previously documented Europe 2 lineage sequences in the maximum likelihood analysis (Fig. 1).
CCHFV isolates across Africa and Eurasia are known to exhibit significant sequence diversity and phylogenetically clustered into distinct lineages or genotypes, commonly designated with Roman numerals and geographic location (Lukashev et al. 2016). In Turkey, Europe 1 (clade V) and Europe 2 (genotype VI) lineages have been reported from ticks as well as symptomatic humans (Midilli et al. 2009, Ozkaya et al. 2010, Gargili et al. 2011, Dinçer et al. 2017). CCHFV Europe 2 strains (also called previously as the AP92-like, named after the initial isolate) have received particular interest due to rare detection in human infections, mostly presenting with mild symptoms and low mortality.
Originally identified in R. bursa ticks from Greece (Papadopoulos and Koptopoulos 1980), this CCHFV lineage has also been reported from other Balkan countries (Albania, Bulgaria and Kosovo), Algeria, and Iran (Papa et al. 2014, Sherifi et al. 2014, Kautman et al. 2016, Panayotova et al. 2016). In Turkey, it has been identified in several Rhipicephalus and Hyalomma spp. Ticks, including R. bursa from various regions, with in silico evidence for recombination among strains (Gargili et al. 2011, Dinçer et al. 2017, Ergünay et al. 2020a). However, this is its initial documentation from central Anatolia, especially in Konya province where other closely related orthonairoviruses, including Tamdy virus and the novel Meram virus, were previously reported (Brinkmann et al. 2018, Ergünay et al. 2020a), with further possibilities for genetic exchange.
The clinical presentation and impact of infections with this lineage also require elucidation and can be facilitated with specific nucleic acid detection that can be included in the work-up of suspected cases presenting with tick bites in CCHFV-endemic regions (Ergünay et al. 2020b). In 2021, the International Committee on Taxonomy of Viruses (ICTV) officially reclassified the Europe 2 (genotype VI) viruses as a separate species (Congoid orthonairovirus) and renamed the AP92 strain as Aigai virus (Kuhn et al. 2021), justifying the need for adequate tools to diagnose probable cases and monitor spread.
We further identified JMTV sequences in the study cohort with identical prevalence as CCHFV, in R. bursa/R. turanicus ticks (Table 4). The sequences were highly similar and phylogenetically related to the viruses from Turkey and Balkans (Fig. 2). Initially characterized in Rhipicephalus microplus ticks from China, the genome of JMTV comprises four positive-sense RNA segments, where two segments encoding for the nonstructural proteins are related to flavivirus proteins (Qin et al. 2014).
JMTV and related viruses are globally distributed and detected in several countries, including China, Kosovo, Romania, Turkey, Uganda, North America, Brazil, French Antilles, and Trinidad and Tobago (Ladner et al. 2016, Emmerich et al. 2018, de Souza et al. 2018, Dinçer et al. 2019, Jia et al. 2019, Pascoal et al. 2019, Sameroff et al. 2019, Temmam et al. 2019, Guo et al. 2020, Vandegrift et al. 2020, Yu et al. 2020, Bratuleanu et al. 2021, Kholodilov et al. 2021). In addition to ticks and mosquitoes, JMTVs have been identified in cattle, rodents, and primates, and recently in bats (Qin et al. 2014, Ladner et al. 2016, Emmerich et al. 2018, de Souza et al. 2018, Jia et al. 2019, Yu et al. 2020), suggesting replication in a broad range of hosts.
Moreover, several phylogenetically distinct viruses with similar genome structure, such as Alongshan virus and Yanggou tick virus that are sometimes referred as JMTV-like or the Jingmen virus group, have been identified in similar hosts with a broad distribution (Shi et al. 2015, Kholodilov et al. 2020, 2021). We have previously reported JMTVs from various species of Rhipicephalus and Haemaphysalis spp. Ticks, including the currently identified species, as well as Hyalomma marginatum, the main CCHFV vector in Turkey (Dinçer et al. 2019). However, infected ticks from central Anatolia were initially documented in this study. It appears that JMTVs are widely distributed in all major tick species in Anatolia and eastern Thrace.
Within the Jingmen virus group, JMTV and Alongshan virus are reported as causative agents of human disease, associated with tick-borne infections presenting with mild-to-severe febrile conditions without any differentiating feature from infections by other tick-borne pathogens (Jia et al. 2019, Wang et al. 2019). Furthermore, JMTV detection in blood from CCHFV-infected individuals in Kosovo also indicate human infections potentially transmitted through infected ticks (Emmerich et al. 2018).
We could not previously detect JMTV in human clinical specimens, canine or bovine sera, or tissues from migratory birds (Dinçer et al. 2019). However, considering its ubiquitous distribution further confirmed in this study, JMTV must be sought in individuals with febrile diseases of unknown etiology associated with or without tick bites. Moreover, asymptomatic human exposure should also be screened by serology in regions with documented virus activity.
Finally, we detected TcTV-2 exclusively in D. marginatus ticks from central Anatolia, with an overall prevalence of 1.3% (Tables 3 and 4). Recently classified as the sole member in the newly established Tacheng uukuvirus species (family Phenuiviridae genus Uukuvirus), TcTV-2 was identified during a metagenome investigation of various arthropods in D. marginatus ticks from China (Li et al. 2015).
Follow-up efforts further identified the virus in Dermacentor nuttalli, Dermacentor silvarum, and Hyalomma asiaticum ticks, as well as in a patient with febrile disease (Dong et al. 2021). The presence of virus RNA in patient blood, urine, and throat swab suggested that person-to-person transmission might be possible through droplets or direct contact with body fluids. Further detections in Hyalomma/Dermacentor spp. from Kazakhstan and Dermacentor reticulatus from Romania are documented (Bratuleanu et al. 2021).
Similarly, we initially detected TcTV-2 in a pool of female H. marginatum ticks collected from cattle in the Aegean province of Mugla, and a large portion of the L genome segment encoding for the viral replicase became available (Brinkmann et al. 2018). In this study, we identified TcTV-2 in D. marginatus specimens by targeting the S segment. The sequences formed a well-defined phylogenetically segregated group, sharing a common ancestor with sequences from China and Kazakstan (Fig. 3).
It is interesting that two D. marginatus ticks within a total of ten specimens in the study cohort was positive (Table 3). Detection in the male specimen also suggests probable trans-stadial or horizontal virus transmission among ticks. Very scarce information has so far been available on global epidemiology of TcTV-2. Meanwhile, repeated detection in Anatolia in temporally and spatially separated instances suggest ongoing virus activity, which warrants further investigation in probable vector ticks as well as human infections.
The main limitation of the study stems from the screening by pan-nairovirus and pan-flavivirus assays. Although validated using a number of viruses, these assays are far from being accurate, inevitably prone to bias and with varied sensitivity for individual viruses. Next-generation sequencing (NGS) approaches have been increasingly employed to investigate microbial diversity in ticks and can provide broad-range information on virus genomes (Li et al. 2015, Brinkmann et al. 2018). However, the operational costs of NGS still hampers its widespread use for vector screening. In this study, field-deployable sequencing platforms and assays based on nontraditional detection approaches are likely to help circumvent the current limitations of NGS and facilitate fit-for-purpose virus monitorization in ticks (Myhrvold et al. 2018, Russell et al. 2018).
Another limitation is the cross-sectional specimen acquisition and pooling strategy, yielding an abundant number of specimens from hosts but hampers a thorough assessment of seasonal and temporal aspects of virus activity, as well as viruses in questing ticks. Nevertheless, it provides an efficient approach to screen and confirm virus circulation and probable zoonotic exposure. Efforts for virus isolation and genome characterization are underway.
In conclusion, we detected and characterized recently described tick-borne pathogens, JMTV and TcTV-2, in several regions. These viruses should be screened as potential etiological agents in human infections associated with tick bites. More information on virus distribution and diversity obtained through robust screening approaches are required.
Footnotes
Author Disclosure Statement
The authors have no potential, actual, or financial competing interests to declare.
Funding Information
No official funding was received for the study.