
Abstract
Objective:
We aim to investigate the species composition of ticks and the pathogen characteristics they carry in the Argun port area of the China–Russia border.
Materials and Methods:
Ticks were collected in surrounding grassland, mixed forest land, and other different habitats around the Argun port area at the Sino-Russian Border of Inner Mongolia in China in April 2019. The presence of 16 potential pathogens, including Yersinia Pestis, Francisella tularensis, Coxiella burnetii (Cb), Anaplasma sp. (Ap), spotted fever group rickettsiae (SFG Rk), Borrelia sp. (Bl), Leptospira, Bartonella spp., Babesia, Crimean-Congo hemorrhagic fever virus, tick-borne encephalitis virus, Bhanja virus, West Nile Virus, severe fever with thrombocytopenia syndrome bunyavirus, Hantaan virus, and bocavirus (boca) was analyzed by polymerase chain reaction. The DNA and amino acid sequences of tick-borne pathogens were compared for homology, and the phylogenetic trees were constructed by using Mega and Lasergene software.
Results:
A total of 210 ticks were collected and they belonged to three species: Dermacentor nuttalli, Ixodes persulcatus, and Haemaphysalis verticalis. Among them, 165 (78.57%) ticks tested positive for 5 pathogens, namely Ap, SFG Rk, Cb, Bl, and boca. Fifteen (7.14%) ticks were detected coinfection with two pathogens, and none were coinfected with three or more pathogens.
Conclusion:
This study shows the prevalence of at least five tick-borne pathogens in Argun, and there is a risk of coinfection by two pathogens in one tick. This study reveals the great importance of controlling tick-borne diseases in this region.
Introduction
Ticks, a widespread host of various pathogens, are the second most common vectors of human pathogens worldwide after mosquitoes. They are obligate nonpermanent ectoparasites of terrestrial vertebrates that cause harm to their host through direct bites or the spreading pathogens (Cao et al., 1999; Colwell et al., 2011; Yin et al., 2019; Zhu et al., 2016). Tick-borne diseases are zoonotic diseases and also natural epidemic diseases. These zoonoses are recognized as emerging or re-emerging infectious diseases spread by infected ticks (Liu et al., 2015). Recently, the types of human-infected tick-borne diseases and the number of cases reported in China have been increasing (Fang et al., 2015). An epidemiological survey on ticks shows that a variety of ticks can obtain pathogens from their host, including 83 species of viruses, 31 species of bacteria, 20 species of rickettsiae, 18 species of spirochetes, 32 species of protozoa, 1 species of chlamydia, 1 species of bartonella, and 2 species of nematodes (Zhang and Zhao, 2005).
Anaplasmosis is a type of tick-borne infectious disease that can cause acute and chronic infection of humans and animals, and its pathogens include Anaplasma marginale, Anaplasma centrale, Anaplasma phagocytophilum, Anaplasma bovis, Anaplasma ovis, and Anaplasma capra (Li et al., 2015). In 1983, Ma et al. first discovered anaplasmosis in sheep in Xinjiang, China. Later, A. ovis was reported in Liaoning, Inner Mongolia, Gansu, Shaanxi, Ningxia, Qinghai, Henan, Shandong, Sichuan, and other regions of China (Song et al., 2011). Studies found that A. ovis was mainly transmitted by Hyalomma asiaticum, Dermacentor nuttalli, Rhipicephalus pumilio, Dermacentor niveus, and Dermacentor marginatus (Dai et al., 2015; Xu et al., 2018).
Tick-borne spotted fever group (SFG) Rickettsia is caused by obligate intracellular Rickettsia Gram negative bacteria, and more than 10 S. rickettsiae species have been identified internationally (Ye et al., 2008). Ticks can transmit microorganisms to humans and animals through bites. Heilongjiang, Inner Mongolia, and Xinjiang were identified as epidemic area of SFG Rickettsiae (SFG Rk) in China (Zhang, 1999).
Q fever is caused by Coxiella burnetii (Cb), an intracellular organism whose hosts include birds, arthropods, and wild and domestic mammals (Masala et al., 2004). Ticks were confirmed to be the most common carrier of Cb (Seo et al., 2016). Lyme disease is a natural-focal disease caused by different genotypes of Borrelia sp. (Bl) and is transmitted by tick bites in humans and animals (Steere, 1989). Since Lyme disease was first investigated in 1986, cases have been reported in 29 provinces in China. Among them, Heilongjiang, Inner Mongolia, Jilin province, and other northern regions are dominated by Borrelia garinii and B. afzelii (Yu et al., 2015).
The Parvoviridae family consists of eight genera of small viruses, including Amdoparvovirus, Aveparvovirus, Bocaparvovirus, Copiparvovirus, Dependoparvovirus, Erythroparvovirus, Protoparvovirus, and Tetraparvovirus. Porcine bocavirus, bat bocavirus, and rodent bocavirus have been detected and reported in China (Cotmore et al., 2014; Jiang et al., 2017a; Xu et al., 2018; Zhu et al., 2019). One case of tick bocavirus has also been reported (Wang et al., 2017).
This study aims to investigate the prevalence of ticks infected with pathogens, including Anaplasma sp. (Ap), SFG Rk, Cb, Bl, Leptospira (Ls), Bartonella spp. (Bt), Francisella tularensis (Ft), Babesia, Crimean-Congo hemorrhagic fever virus (CCHF), tick-borne encephalitis virus (TBE), Bhanja virus (BHAV), West Nile Virus (WNV), severe fever with thrombocytopenia syndrome bunyavirus (SFTSV), Hantaan virus (HTNV), and boca, and the presence of coinfections with these pathogens. The study also characterizes molecular signatures of tick-borne pathogens detected in the Argun port area.
Materials and Methods
Ticks collection and morphological identification
Ticks were collected from various habitats, such as grass and shrubs, in the Argun port area, China. Free ticks were captured using cloth-dragging method (Yang et al., 2018). After identifying their species and sex, the ticks were immediately stored in 70% ethanol and transported to the laboratory for further processing.
Nucleic acid extraction
Ticks were washed once in 70% ethanol for 5 min and then twice in distilled water for 5 min each. Each tick was ground individually using a tissue sample grinder. Total genomic DNA and/or RNA were extracted from the ticks using AllPrep DNA/RNA Mini Kit (QIAGEN Bio Co., Ltd., Shenzhen, China), according to the manufacturer's instructions. DNA was eluted with 100 μL of buffer and stored at −20°C, RNA was reverse transcribed into cDNA using PrimeScript RTase (Takara Biotech [Beijing] Co., Ltd.), then stored at −80°C. The final concentration of the template for each polymerase chain reaction (PCR) was <200 ng.
Molecular analysis of pathogens
The presence of 16 potential tick-borne pathogens were screened with PCR methods. The target genes were amplified using PCR or nested PCR, and the real-time PCR (RT PCR) was used for the detection of the following pathogens: CCHF, TBE, BHAV, WNV, and SFTSV (Table 1).
Polymerase Chain Reaction Assays Used for Tick-Borne Pathogens Screening
BHAV, Bhanja virus; CCHF, Crimean-Congo hemorrhagic fever virus; HTNV, Hantaan virus; HTV; NS1; PCR, polymerase chain reaction; RT PCR, real-time polymerase chain reaction; SEOV; SFG, spotted fever group; SFTSV, syndrome bunyavirus; TBE, tick-borne encephalitis virus; WNV, West Nile Virus.
Composition of Tick Species in Argun Port Area
Reactions were performed using Applied Biosystems GeneAmp PCR System 9700 and 7500 Fast RT PCR Systems (Courtabouef, France). Tick DNA or RNA was used as templates for PCR, and negative and positive controls were also included. PCR products were electrophoresed in 2% agarose gel and visualized under ultraviolet light. A 50-bp DNA ladder was used as a standard marker.
Statistical analysis
SPSS Statistics for Windows, Version 19.0 (IBM Corp., Armonk, NY) was used for data analysis. Fisher exact test was used to analyze the detection rate of pathogens in collected samples (p < 0.05). A univariate analysis chi-squared test was used to analyze the infection rates of ticks of distinct species and sexes. A univariable nonconditional logistic regression model was employed to analyze factors that may influence the pathogens prevalence. Two-tailed p < 0.05 was the criterion used to determine a statistically significant difference.
DNA sequencing and phylogenetic analysis
A selected number of PCR-positive products were purified using the Omega Gel extraction kit (BioTek Instruments, Inc., Winooski, VT). The purified DNA fragments were sequenced by Thermo Fisher Scientific (China). The sequencing data were aligned using MEGA X, and all sequences were assembled and compared with similar sequences retrieved from the GeneBank nucleotide database using BLAST. MEGA software was used to perform sequence analysis, and Kiruma two parameter model was used for calculation. MrBayes software was used for the best model performing Bayesian analysis (BI). Meanwhile, a neighbor-joining tree of amino acid sequences was constructed using the MEGA software Poisson correction model.
Region of the current study
Argun area is the northernmost border city in China with geographical coordinates of 50°01 “∼53°26” north and 119°07 “∼121°49” east. It is the third largest subdivision of the country, spanning about 28,000 km2. Shiwei and Heishantou are the regions of naturalistic importance located at the Sino-Russian borders (Fig. 1). The average annual temperature in the urban area is between −2.0°C and 3.0°C, and the annual rainfall is 200 to 280 mm.
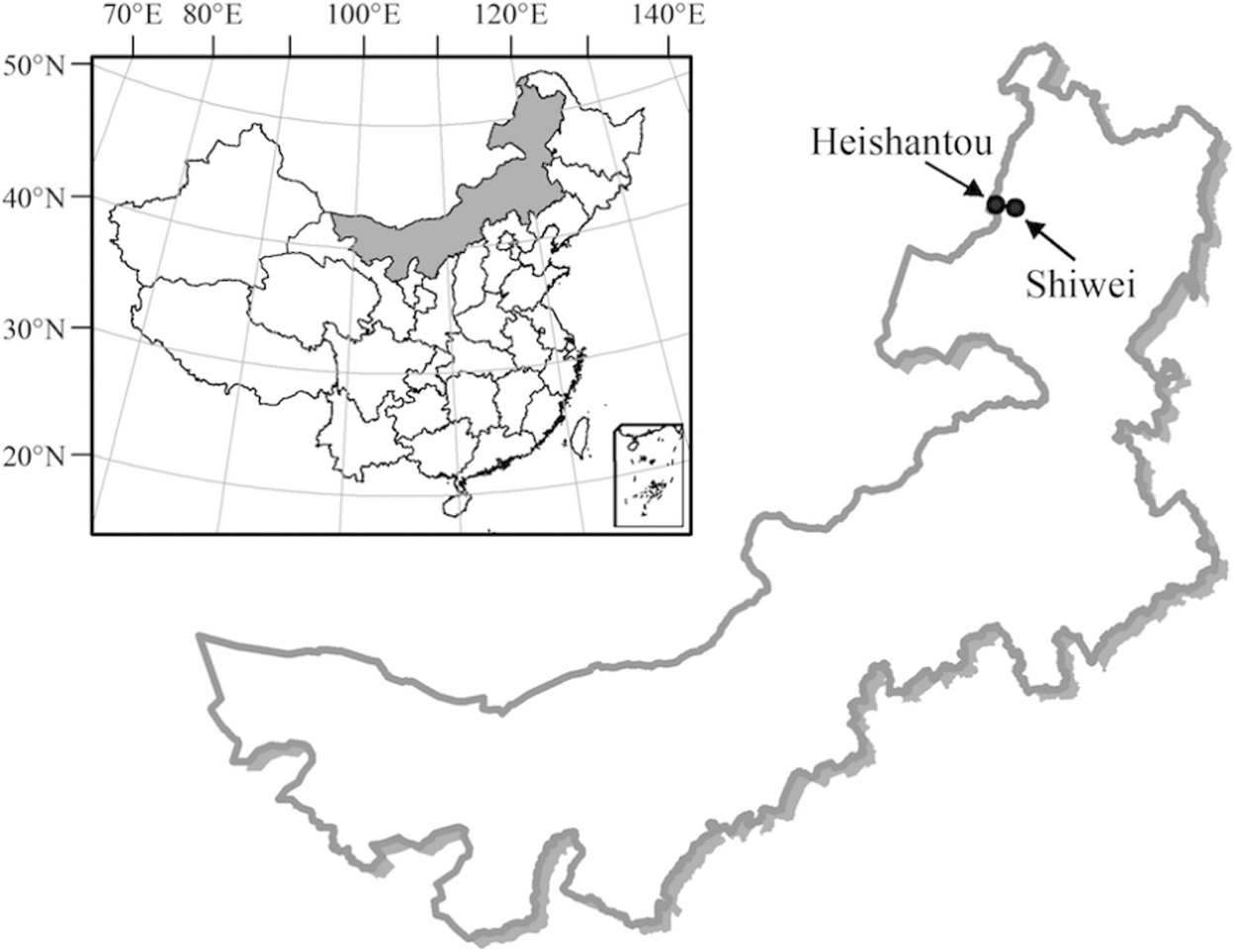
The sites of tick collection at the Sino-Russian borders.
Ethical consideration
Local Ethics Committee of the Institute of Health Inspection and Quarantine, Chinese Academy of Inspection and Quarantine (CAIQIHIQ) authorized the conduct of this research in 2019. Ethical approval number was CIAQIHIQ2019-A0004. All tick samples were performed by following the guidelines of CAIQIHIQ.
Results
Tick identification
A total of 210 ticks belonging to three species of one family were collected from Argun port area in April 2019 (Table 1). Of them, 114 were female ticks (54.29%), and 96 were male (45.71%). All tick samples were identified at the species level. D. nuttalli was the predominate tick species, with a population of 179 (85.24%), including 104 females (58.10%) and 75 males (41.90%); followed by Ixodes persulcatus (23, 10.95%), consisting of 10 female ticks (43.48%) and 13 male ticks (56.53%). Haemaphysalis verticalis was the minority tick (3.81%, all male).
Detection of tick pathogens
Samples were considered positive when PCR yielded fragments with the expected length of the pathogens, or fluorescence PCR presented a typical amplification curve. Five pathogens (Ap, SFG Rk, Cb, Bl, boca) were detected positive in 165 tick samples, including 143 ticks of SFG Rk, 11 of Ap, 3 Cb, 2 f Bl, and 6 boca. The detailed results are summarized as following.
A DNA fragment of a partial ompA gene of SFG Rk was detected positive by PCR in 143 of 210 ticks (68.10%). The overall prevalence of SFG Rk was significantly highest compared with other pathogens. The highest Infection rate of SFG Rk was in I. persulcatus (n = 18, 78.26%), followed by that in D. nuttalli (n = 121, 67.60%), then in H. verticalis (n = 4, 4/8) (Table 3).
Infection Rate of Different Pathogens in Ticks and Logistic Regression Analysis
CI, confidence interval; OR, odds ratio; PE, parameter estimate; SE, standard error.
Nested PCR was used to detect partial 16S rRNA gene of AP, with a total of 11 (5.24%) positive samples. The positive infection rate of Ap in D. nuttalli ticks (n = 10, 5.59%) was little higher than that in I. persulcatus (n = 1, 4.35%), but none of H. verticalis was found infected by Ap (Table 3).
Nested PCR for a partial outer membrane protein-coding gene (Com1 gene) detected Cb DNA fragments in 3 of 179 ticks (1.68%). The results showed that all positive detections were in female D. nuttalli. By contrast, I. persulcatus and H. verticalis were negative for Cb DNA (Table 3).
DNA fragments of Bl were detected by nested PCR in a portion of the 5s–23s rRNA intergenic region in two tick samples of I. persulcatus (8.70%). None of D. nuttalli and H. verticalis tested positive for Bl.
The extracted Boca RNA was reverse transcribed into cDNA for detection. Nested PCR detected six ticks (26.09%) of I. persulcatus positive for the NS1 fragment, most of them were males, and only one was female. Neither D. nuttalli nor H. verticalis was infected by Boca.
In addition, 15 samples of the 210 ticks (7.14%) were coinfected with 2 pathogens, of which 5 I. persulcatus (21.74%) were infected with SFG Rk and Boca; 2 I. persulcatus (8.70%) coinfected with SFG Rk and Bl; 2 D. nuttalli (1.12%) coinfected with SFG Rk and Cb; 6 D. nuttalli (3.35%) coinfected with SFG Rk and Ap. None of H. verticalis were detected as coinfected, and no ticks were coinfected with three or more pathogens. The detected results are listed in Table 3.
Sequencing and phylogenetic analysis
DNA fragments of the ompA gene of four Rickettsia isolated in Argun were sequenced. After blast sequences alignment, the nucleotide sequences of the genomic coding region of ompA gene were aligned using Clustal-X 2.0.11. Among which, ER-169 and ER-130 belonged to the same branch, indicating that the two SFG Rk isolated in Argun were closely related. In addition, ER-169, ER-130, and ER-005 belonged to the same cluster as the Rickettsia heilongjiangensi (Genebank ID: AP019865) isolated in Japan, indicating that ER-169, ER-130, and ER-005 were closely related to Rickettsia heilongjiangensi. ER-30 has close homology to Rickettsia parkeri (Genebank ID: CP040325) and belongs to the same branch (as shown in Fig. 2). Analysis of the ompA gene sequences revealed the presence at least two species of Rickettsia, Rickettsia heilongjiangensis, and R. parkeri.
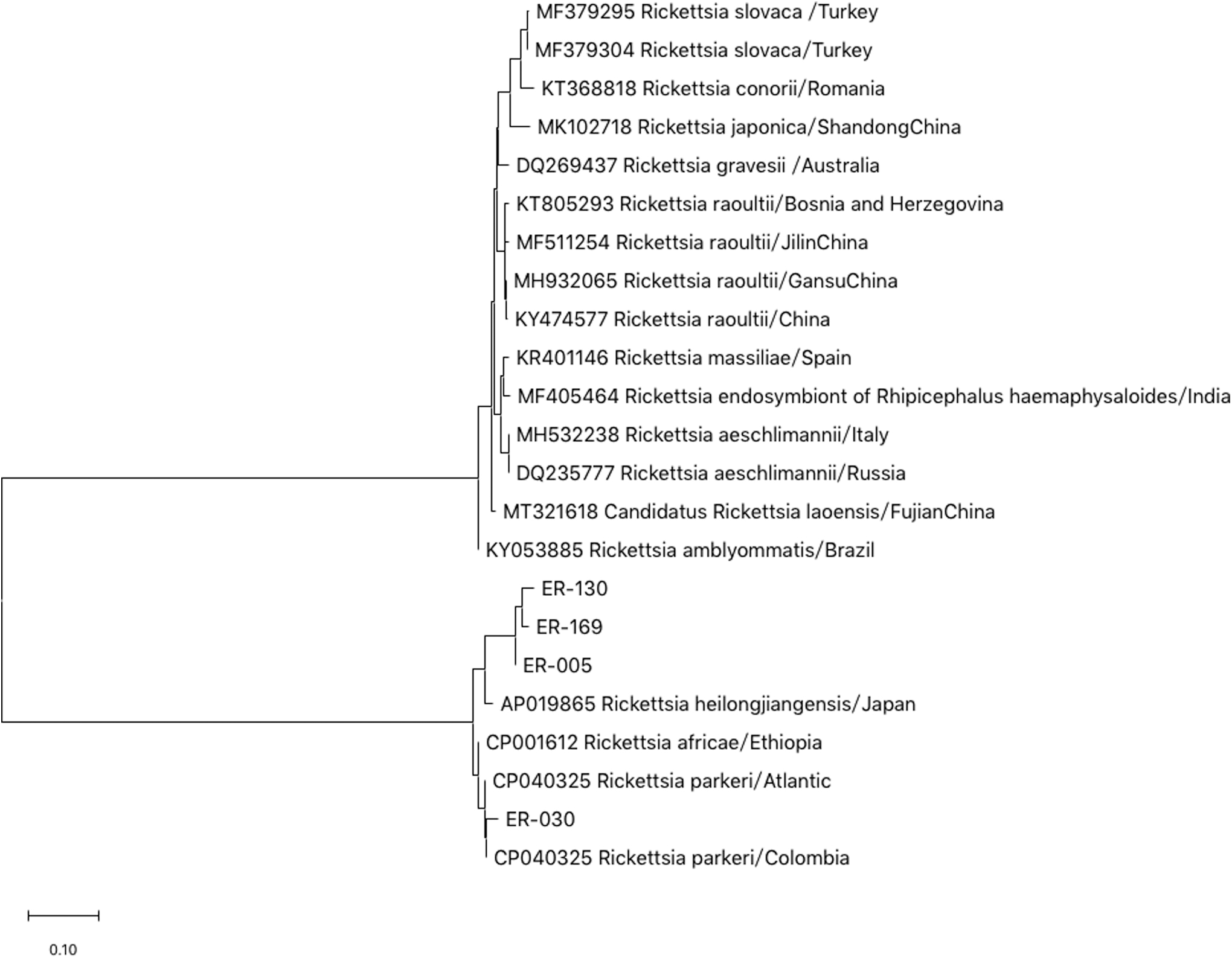
Phylogenetic tree of Rickettsia based on the ompA gene. The trees were calculated by the neighbor-joining method using MEGA X software. Values of the bootstrap support of the particular branching calculated for 1000 replicated are indicated at the nodes. The variant sequences obtained in the study are designated by accession number and species.
Partial 16S rRNA gene sequences of Ap from GeneBank entries and three strains isolated in Argun were selected for the phylogenetic analysis. Phylogenetic trees were constructed using Anaplasma 16S rRNA gene fragments in MEGA X with the neighbor-joining method (1000 bootstrap replications). Bootstrap values (>70%) were shown at the branches. As evident from Fig. 3, analysis of the ER028 and ER099 sequences revealed the presence of one species A. ovis. ER028 and ER099 were clustered with A. ovis (ID: EP587237) found in Guangxi, China. ER100 aligned with the 16 selected sequences but failed to cluster with known Ap.
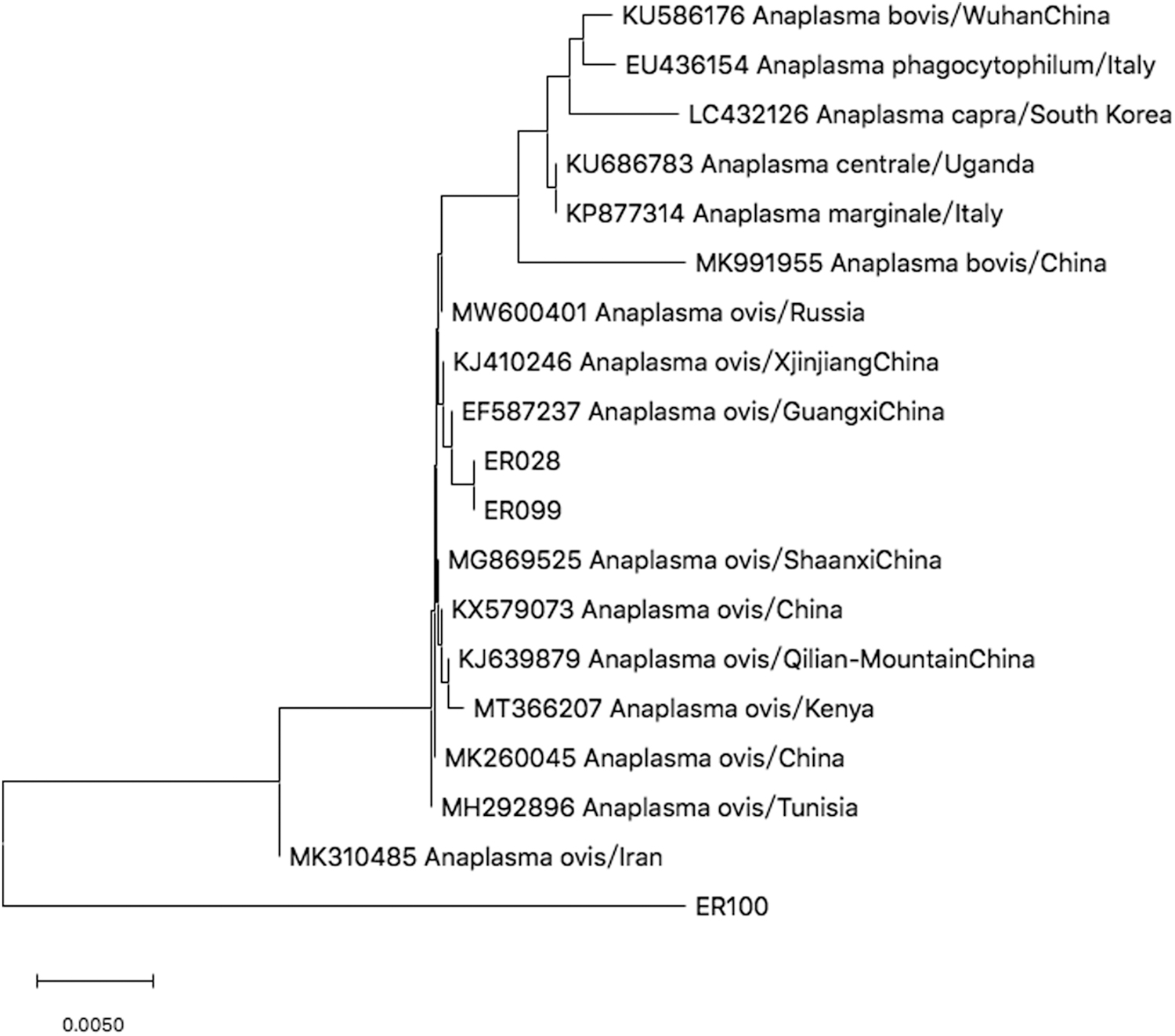
Phylogenetic tree of Anaplasma ovis base on Partial 16S rRNA gene. The trees were calculated by the neighbor-joining method using MEGA X software. Values of the bootstrap support of the particular branching calculated for 1000 replicated are indicated at the nodes. The variant sequences obtained in the study are designated by accession number and species.
Two DNA sequences of Bl 5s–23s rRNA were obtained by sequencing. Borrelia burgdorferi (ID: L42877) had the highest homology with ER-137. ER-168 had the highest homology with the sequence of B. garinii isolated from I. persulcatus in Russia (ID:CP003201). Nineteen submitted gene sequences were selected from GeneBank to construct a neighbor-joining tree (as shown in Fig. 4). Analysis of the partial 5s–23s rRNA intergenic region revealed the presence of B. burgdorferi and B. garinii.
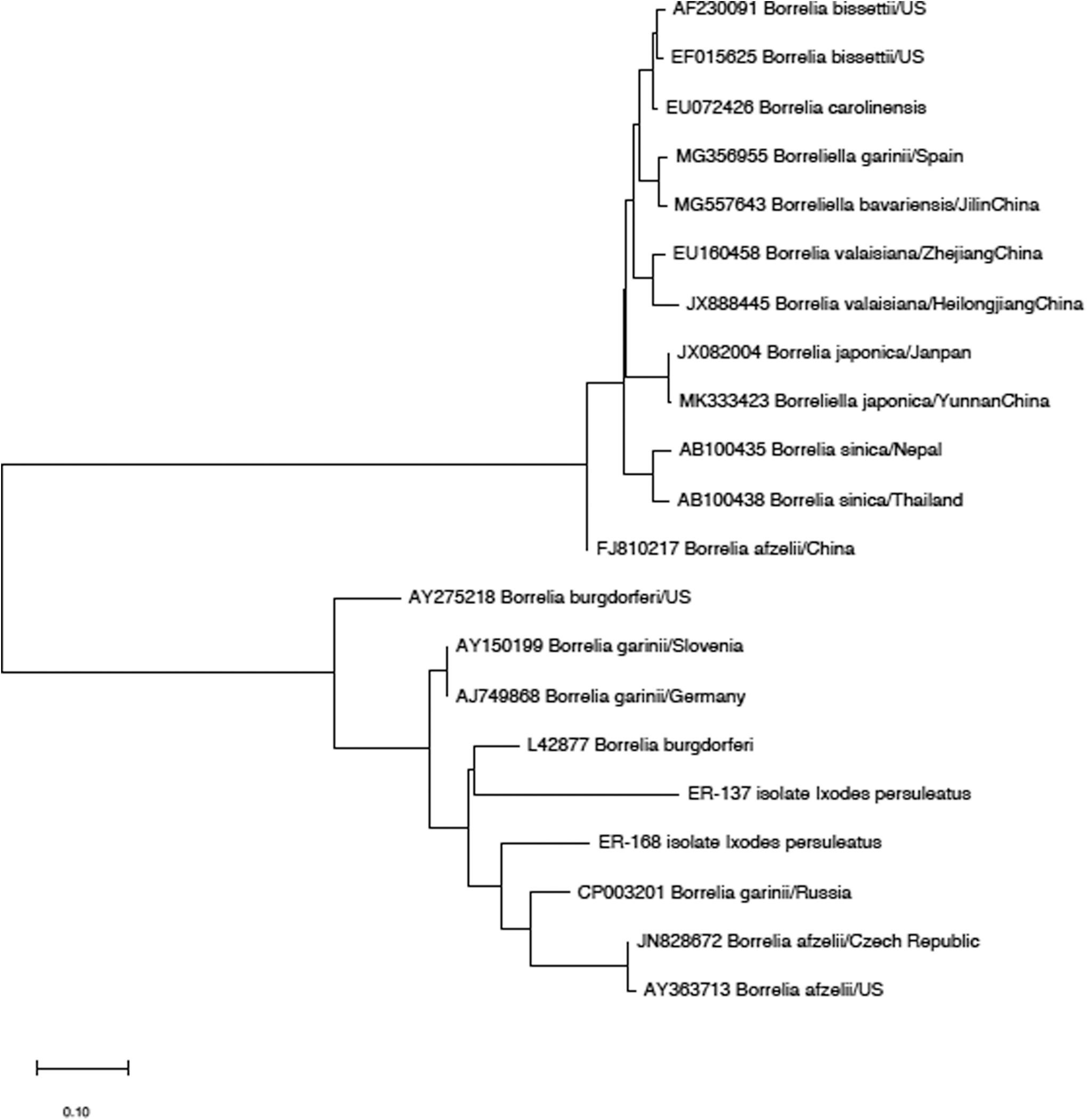
Phylogenetic tree of Borrelia sp. base on 5s–23s rRNA sequences. The trees were calculated by the neighbor-joining method using MEGA X software. Values of the bootstrap support of the particular branching calculated for 1000 replicated are indicated at the nodes. The variant sequences obtained in the study are designated by accession number and species.
Sequence analysis of the Com1 gene revealed the presence of Cb. Three DNA sequences of omp gene of Cb were obtained by sequencing. The sequences of ER-27, ER-110, and ER-203 had the highest homology with the sequence of the Iran strain (ID: JX131364). Eleven submitted gene sequences were selected from GeneBank to a construct a neighbor-joining tree (as shown in Fig. 5).
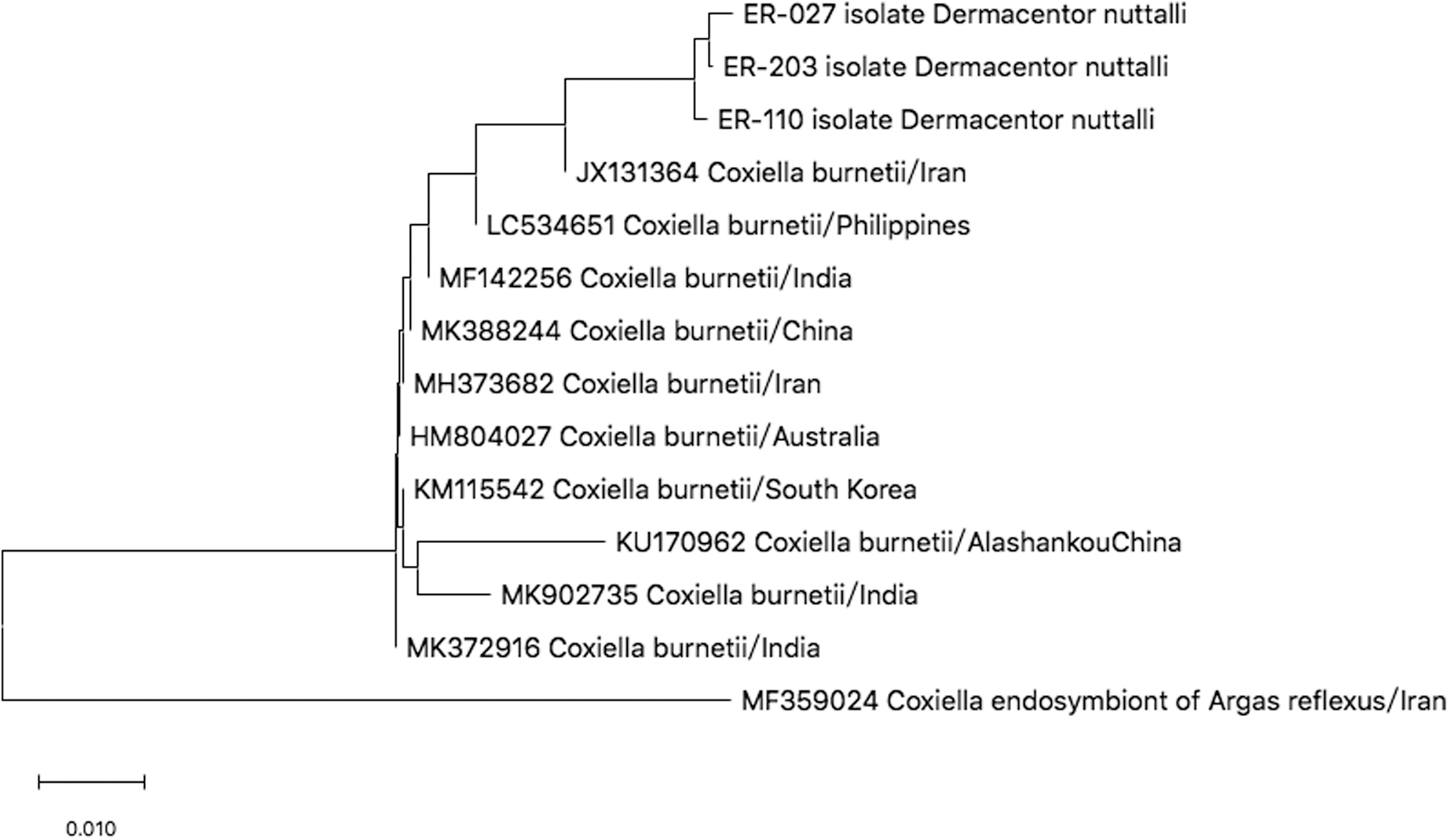
Phylogenetic tree of Coxiella burnetii with Com1 gene. The trees were calculated by the neighbor-joining method using MEGA X software. Values of the bootstrap support of the particular branching calculated for 1000 replicated are indicated at the nodes. The variant sequences obtained in the study are designated by accession number and species.
Two DNA fragments of boca obtained were sequenced. Sequence analysis revealed the presence of the boca. Among them, ER-25 strain was highly homologous with the Chinese strain of Bocaparvovirus (ID: MF136076) isolated from Meriones erythrourus in Alataw Pass, Xinjiang Autonomous Region. Strain ER-74 was closely related to Rodent bocavirus (ID: KY927870), which was isolated from Rattus flavipectus in China. Eleven submitted gene sequences from Genebank were selected to construct the neighbor-joining tree (Fig. 6).
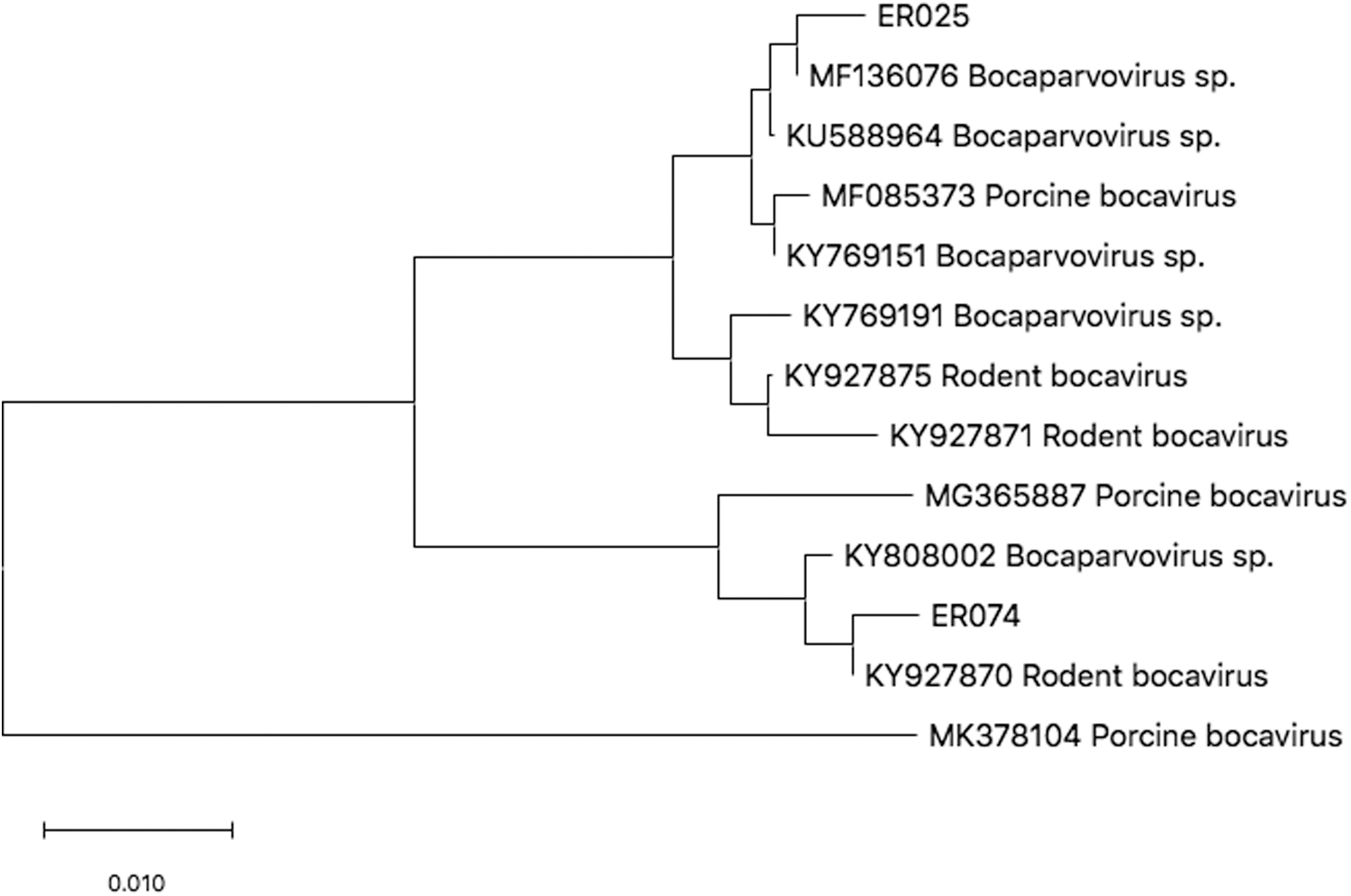
Phylogenetic tree of bocavirus. The trees were calculated by the neighbor-joining method using MEGA X software. Values of the bootstrap support of the particular branching calculated for 1000 replicated are indicated at the nodes. The variant sequences obtained in the study are designated by accession number and species.
Discussion
There are currently two national-level ports in the Argun region China. Despite the large trading volume, there is still little information about the tick epidemic. To the best of our knowledge, there are four species of ticks (Dermacentor nuttalli, Dermacentor silvarum, Haemaphysalis concinna, and I. persulcatus) at the Argun port on the Sino-Russian border and in adjacent forest areas in China (Wang et al., 2017; Zhu et al., 2015). Only the following three species of ticks were collected in our survey, namely D. nuttalli, H. verticalis, and I. persulcatus. The results of this study were somewhat different from those of previous surveys, it may be due to the limitation of sampling sites, which can only reflect the population structure of ticks in some local areas.
This study demonstrated common tick species in the Argun port area of China and the presence of A. ovis, SFG Rk, Cb, Bl (B. burgdorferi and B. garinii), and boca in collected ticks. Other pathogens, such as Ls, Bt, Ft, Babesia, CCHF, TBE, BHAV, WNV, SFTSV, and HTNV, were not found in our study.
Rickettsia spp. was widely distributed, and there were at least 10 species of Rickettsia found in China (Zhang et al., 2007). SFG Rk was one of the prevalent Rk spp. and has been isolated from ticks in Xinjiang, Inner Mongolia, Heilongjiang, and other provinces of China, among which I. persulcatus had the highest infection rate (Sun et al., 2007a). This study found that the three tick species were all with high infection rate of SFG Rk, as I. persulcatus (18/23, 78.26%), D. nuttalli (121/179, 67.60%), and H. verticalis (4/8, 50%). These findings suggested that there was no apparent interspecies difference in SFG Rk infection rate in ticks. We speculated that SFG Rk could be carried and transmitted in different tick species. Based on nucleic acid sequences alignment on NCBI, it was demonstrated that the ticks captured in our investigation were infected with Rickettsia raoultii.
A previous study found that Ap was a pathogen with a higher infection rate than other tick-borne diseases (Cheng et al., 2019). Ap is commonly infected in I. persulcatus and D. nuttalli, which was consistent with our study. We found that 11 ticks were infected with Ap, the second common pathogen after SFG Rk. Infected ticks detected in this survey included one I. persulcatus and 10 D. nuttalli, but none of the pathogen-infected H. verticalis was detected. Ap had not been reported in the same study area, but it had been detected elsewhere in Inner Mongolia, such as Manchuria.
According to surveys, the Lyme disease spirochete was first isolated from I. persulcatus in China in 1987, and I. persulcatus was the primary biological vector of B. burgdorferi in northern China (Wan et al., 1998; Zhang et al., 1989). Since the 1986 Lyme disease survey in China, relevant findings have been reported in 29 provinces. There are at least five genotypes of B. burgdorferi in China: B. burgdorferi B. garinii, B. afzelii, B. yangze sp., and B. sinica. Among them, B. garinii and B. afzelii were the main strains in northern provinces such as Heilongjiang, Inner Mongolia, and Jilin, and B. garinii predominant was the dominant strain in China mainly in northern areas (Yang et al., 2015; Zhang et al., 2015).
The main biological vector of B. burgdorferi in northern China was I. persulcatus. In this study, we detected B. burgdorferi and B. garinii in I. persulcatus but not in other two tick species. According to previous studies, the occurrence of Lyme disease was associated with a specific tick I. persulcatus, which was the main transmitter in northern China (Wan et al., 1998). The present findings confirmed that I. persulcatus was the main vector of Lyme disease in the Argun port area.
Several tick samples collected in Xinjiang province tested positive for Cb (Jiang et al., 2017b; Wang et al., 2017). It is well known that Q fever is caused by Cb. Previous studies showed that Q fever outbreaks had occurred in China, including Inner Mongolia, Sichuan, Xinjiang, Yunnan, and Tibet province (Yu, 2000). More than 40 species of tick may be potential hosts for Cb, and Cb can easily infect ticks at any stage of tick development (Balashov and Daiter, 1973; Norlander, 2000). According to this investigation, three ticks of D. nuttalli (1.68%) were infected by Cb, whereas neither I. persulcatus nor H. verticalis tested positive for Cb.
Previous finding suggested that Boca detected in ticks should be classified as a new member of species Ungulate bocaparvovirus 4 (Wang et al., 2017). Only one case of tick Bocavirus has been reported, which was found in I. persulcatus and D. nuttalli. In this study, Boca infection was only found in I. persulcatus, suggesting that I. persulcatus might be the main transmission route of Boca in the Argun area. The genetic and host diversity of Boca is still poorly understood. More than one tick infection with Boca was detected in the Argun region, revealing the risk of Boca transmission by tick in the area.
We found several cases of coinfection in I. persulcatus and D. nuttalli, including six cases of coinfection of A. ovis and SFG Rk, two cases of coinfection of SFG Rk and Cb, two cases of coinfection of SFG Rk and Bl (B. burgdorferi and B. garinii), and five cases of coinfection of SFG Rk and boca, respectively. Tick coinfection means that a single tick is infected with two or more pathogens, significantly increasing the risk of tick bite. Forms of coinfection include interspecific infection and intraspecific infection between different genotypes. Tick species have some influence on coinfections.
Investigations had found that they were mostly concentrated in I. scapularis, I. pacificus, I. ricinus, and I. persulcatus of the family I. scapularis (Bao, 2007). Currently, most studies on coinfections with tick-borne pathogens have focused on B. burgdorferi, Babesia parvum, Ehrlichia, and A. phagocytophilum. Coinfection of Rickettsia with other pathogens, such as Bl or Ap, was often found in ticks and humans (Satta et al., 2011; Socolovschi et al., 2012). Coinfection of ticks with pathogens varies widely among in different literature reports, and has significant regional variations.
The results of the molecular epidemiological studies indicated that I. persulcatus and D. nuttalli had the possibility of multiple infections in the study region. Differences infection rate were observed between I. persulcatus and D. nuttalli. I. persulcatus has been shown to be naturally infected by a variety of pathogens, including viruses, bacteria, rickettsiae, spirochetes, and at least 40% of I. persulcatus individuals have coinfections (Sun et al., 2007b). Therefore, I. persulcatus is a high-risk tick species that may carry multiple pathogens, which should be given sufficient attention.
Although there have been few previous reports of coinfection in D. nuttalli, it was worth noting that several pathogens, including A. ovis, SFG Rk, and Cb, were detected in D. nuttalli in the port area of Argun in this investigation. More importantly, it contained two cases of coinfections with SFG Rk and Cb and six cases of coinfections with SFG Rk and Ap, which had not been reported in this region. This result highlights that D. nuttalli may be a host of multiple pathogens and a major transmitter of tick-borne diseases and is an important tick species with coinfection in this region. Owing to the diversity of pathogens that vector ticks can transmit and the complexity of epidemic links, the number of coinfected tick-borne diseases is increasing, and the probability of coinfections between host animals and humans is also increasing. However, tick coinfection cases were rarely reported in inner Mongolia.
Although the coinfection in H. verticalis has not been detected, the possibility of coinfection remains uncertain. This study found that the coinfection of pathogens in tick-borne diseases was relatively common, which might lead to difficulties in diagnosing and treating of infectious diseases.
There are some limitations to this study. First, the ticks we studied were collected only on the port area in the Argun region (China), there were no data from the Russia side. Second, the period of our investigation was relatively short, mainly in April, and there might be limitations to tick species. Third, the limited species and uneven distribution of the numbers of ticks we captured might not accurately reflect the severity of tick-borne diseases.
Owing to the development of natural resources, national defense construction, and tourism, human access to natural epidemic foci has increased. Human activities not only affect the number and species of arthropods and their hosts in the epidemic foci, but also change the structure of the foci. Humans enter the epidemic foci and have more opportunities to come to contact with arthropods, so the probability of human infection with zoonotic diseases increases. At the same time, the chance of coinfection with pathogens increases. These results contributed to a deeper understanding to the tick-borne diseases in the Argun area and provide valuable data for epidemiological studies of zoonotic diseases. In addition, further studies should be conducted to investigate the eco-epidemiological cycle of pathogens detected in this area and to identify risk factors for tick-borne diseases infection. In summary, attentions should be paid to detecting vector ticks and monitoring pathogens in and around the Argun port areas.
Conclusion
Two hundred ten ticks were collected in this investigation, including 85.24% Dermacentor nuttalli, 10.95% I. persulcatus, and 3.81% H. verticalis. D. nuttalli was the dominant species in this area. Among the presence of 16 potential pathogens, nucleic acids of Ap, SFG rk, Cb, Bl (B. burgdorferi and B. garinii), and Boca were detected from these tick samples, with a positive rate of 5.24% for Ap, 68.10% for SFG Rk, 1.43% for Cb, 1% for Bl and 2.86% for Boca, respectively. It is worth noting that 15 samples of ticks (7.14%) detected a combined infection of the 2 pathogens. Among them, the coinfection rates of Ap and SFG Rk, SFG Rk and Cb, SFG Rk and Bl, and SFG Rk and Boca coinfection rates were 2.86%, 0.95%, 0.95%, and 3.03%, respectively. Meanwhile, I. persulcatus was a tick vector with a higher coinfection rate (30.43%) than that of the dominant tick species D. nuttalli (4.46%).
This study shows that tick-borne diseases are prevalent in Argun, and there is a risk of coinfection of ticks with multiple pathogens. The study also revealed the prevalence of tick-borne diseases and provided a basis for preventing and controlling the Argun port area in China.
Footnotes
Acknowledgments
We sincerely thank Prof. Yawei Zhang and Kaiyong Zou (Chinese National Clinical Research Center for Cancer) for assistance with this article.
Data Availability
The draft genome assemblies reported here are available in GeneBank under the accession numbers, and all raw sequencing reads have been deposited in the NCBI under the BioProject number and are available in the Sequence Read Archive.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by: National key research and development plan (2018YFF0214901); The Ministry of Science and Technology of the People’s Republic of China (2018ZX10101001-004-002); The Chinese Academy of Inspection and Quarantine (2017JK008).