
Abstract
Background:
Dengue virus (DENV) can be divided into four serotypes—DENV-1, DENV-2, DENV-3, and DENV-4. In humans, infection leads to dengue fever (DF), dengue hemorrhagic fever, and dengue shock syndrome, both widely prevalent in tropical and subtropical regions. In 2019, a severe outbreak of DF occurred in Xishuangbanna, Yunnan province.
Objective:
To investigate the etiology and genotype of the causative agents of this severe dengue outbreak in Xishuangbanna.
Methods:
Between October and November 2019, the sera of patients clinically diagnosed with DF were collected in the first People's Hospital of Xishuangbanna. RNA was extracted from the sera and amplified by RT-PCR with flavivirus primers. Flavivirus-positive sera were then used to inoculate Aedes albopictus cells (C6/36); viral RNA was extracted from these cells, amplified, and sequenced with DENV E gene-specific primers. Sequence splicing and nucleotide homology genetic evolution analysis were carried out by biological software (DNAStar). Unique mutations in the E genes of isolated DENV were analyzed by SWISS-MODEL and PyMOL.
Results:
Of the 60 samples collected from DF patients, 39 tested positively with flavivirus primers. The DENV was isolated from 25 of the 39 positive seras, of which 20 showed cytopathic effects (CPE) and 5 were no CPE. In these 25 isolated nucleic acids, 21 strains of DENV-1, 3 strains of DENV-2, and 1 strain of DENV-3 were identified according to the sequence of E protein. In the four unique mutations (D52, Y149, L312, T386), D52 and Y149 in the E protein of DENV-1 were predicted to be exposed on the surface of the prefusion conformation.
Conclusion:
The 2019 outbreak of DF in Xishuangbanna area of Yunnan Province consists of at least three serotypes of DENV-1, DENV-2, and DENV-3, and the sources of these virus strains are of mixed and complicated origin.
Introduction
Dengue virus (DENV) is mainly transmitted by bites from Aedes albopictus and Ae. aegypti mosquitoes (Zhu et al., 2019). DENV is a spherical particle with a diameter of roughly 50 nm, belonging to the genus Flavivirus, family Flaviviridae (Renner et al., 2021). Each particle contains a unit membrane and an internal core structure. Mature virus particles contain an infectious single-stranded RNA, together with a basic capsid protein. The genome of DENV is about 11 kb in size with a single open reading frame (ORF). Its genome contains a cap structure (m′G5′ppp5′A) at the 5′ end, but no polyadenylate tail at the 3′ end. The genome includes a 5′-UTR, a 3′-UTR, and an ORF. The ORF encodes 3 structural proteins (C, PrM/M, E) and 7 nonstructural proteins (NSl, NS2A, NS2B, NS3, NS4A, NS4B, NS5) with a total of ∼3400 amino acid residues (Gebhard et al., 2011). During the infection, the structural protein envelope (E) of DENV binds host receptor and then initializes the entry process of endocytic pathway. Subsequently, according to the reduced pH in the endosome, a large conformational rearrangement of E from prefusion conformation (dimer) to postfusion conformation (trimer) occurs (Zhang et al., 2013).
According to different epitopes, DENV can be divided into four serotypes: DENV-1, DENV-2, DENV-3, and DENV-4. All four serotypes of DENV can cause varying degrees of clinical symptoms, including generalized fever, mild dengue fever (DF), severe dengue hemorrhagic fever (DHF), and dengue shock syndrome (Jing and Wang, 2019). Infection with DENV causes flu-like symptoms of DF, which can develop into fatal DHF in severe cases. Over the past few decades, the global incidence of DF has increased more than 30-fold, making it one of the most widespread arboviruses (Sanyaolu et al., 2017). The increasing geographical range of the major vector breeding areas of Aedes mosquitoes, increasing international trade, cross-border tourism, deforestation, urbanization, and climate change have also contributed to the spread of DENV (Bouzid et al., 2014). DF mainly occurs in tropical and subtropical countries and regions, but in recent years, local DF outbreaks have also occurred in temperate countries such as France, Portugal, and Japan, indicating that DF is gradually spreading from epidemics to nonepidemics areas (Dehghani and Kassiri, 2021).
Yunnan province is located in the southwest border of China and shares borders with Myanmar, Laos, or Vietnam. Since 2013, there has been a local outbreak of DF in the border areas of southwestern Yunnan, followed by annual local epidemics (Sang et al., 2021). In 2019, a severe outbreak of DF occurred in Xishuangbanna, Yunnan province (Yue et al., 2021). However, the genomic information of DENV is still limited in this outbreak. In this study, we detected and isolated 25 DENVs from the serum in the patients in Xishuangbanna and compared them with the DENVs from previous epidemics in China and other neighboring countries. The results would contribute understanding of the dynamics of DENV infection in Xishuangbanna.
Materials and Methods
Sample collection
From October to November 2019, sera from patients with DF for 1–3 days were collected at the First People's Hospital in Jinghong, Xishuangbanna. The NS1 antigen-positive sera were divided into 2 mL sterile centrifuge tubes, numbered, recorded, deposited in a −20°C vehicle-mounted refrigerator, and transported back to the Key Laboratory of Tropical and Subtropical Animal Viral Diseases, Yunnan Academy of Animal Husbandry and Veterinary Science, and the serum specimens were moved to a −80°C freezer for storage.
RNA extraction and RT-PCR
RNA was extracted according to the Viral RNA Extraction Kit (dp315-r; TIANGEN) method for sera. cDNA was synthesized using random primers P(d)N6 (TaKaRa) and Reverse Transcriptase M-MLV (RNase H-; TaKaRa). The cDNA was subjected to PCR amplification with ExTaq enzyme (TaKaRa); after initial denaturation at 94°C for 5 min, the thermocycling program was 35 cycles of 94°C 30 s, 50°C 30 s, 72°C 60 s, followed by 10 min extension at 72°C. PCR was performed with flavivirus genus primers (FU1: TACAACATGATGGGAAAGAGAGAGAA, cFD2: GTGTCCCAGCCGGCGGTGTCATCAGC) (Kuno, 1998); FU1/cFD2 target gene is polyprotein (POLY). The RT-PCR fragment of 310 bp and PCR amplification products were evaluated by 1.5% agarose gel electrophoresis.
Virus isolation
Ae. albopictus cells (C6/36) were inoculated with sera determined as positive flavivirus with flavivirus genus primer. The input virus was passaged blindly thrice in C6/36 cells. The cytopathic effects (CPE) were observed daily. All the supernatants of cell were stored in the −80°C freezer and used for virus identification.
Sequencing and phylogenetic analysis
Primers were designed with reference to DENV-1, DENV-2, DENV-3, and DENV-4 strains in GenBank (Table 1) to amplify the DENV E gene. RNA was extracted from viral supernatants according to the above method, and cDNA was synthesized. cDNA was amplified by RT-PCR using TaKaRa Ex Taq enzyme in a 50 μL volume, and the PCR products were observed by electrophoresis. The purified DNA fragments were cloned into the pMD19-T vector, and then the ligated products were transformed into Escherichia coli DH5α cells. In the second day, the picked five cloned colonies were subjected to Sanger sequencing by Beijing Qingke Biotechnology Co., Ltd.
Primers for Amplification of E in Dengue Virus
DENV, Dengue virus.
Sequence analysis and comparisons were performed using the DNAStar software package: SeqMan software for sequence splicing, MegAlign for nucleotide and amino acid homology comparisons (Burland, 2000). MEGA6.1 software was applied for phylogenetic analysis (using Neighbor-joining method, bootstrap value of 1000) (Tamura et al., 2013). The structure of E protein in the prefusion conformation and postfusion conformation was predicated by SWISS-MODEL based on 6zqu and 3g7t, respectively, and the protein was visualized by PyMOL (DeLano, 2002; Nayak et al., 2009; Renner et al., 2021; Waterhouse et al., 2018).
Results
Detection and isolation of DENV in samples from sera in the patients with DF
From October to November 2019, 60 sera from DF patients were collected at the First Human Hospital in Jinghong, Xishuangbanna, Yunnan Province, of which 39 sera were positive for RT-PCR amplification of primers of the genus Flavivirus. The 39 positive sera were passaged blindly thrice in C6/36 cells. In the C6/36 culture of the third passage, 20 out of 39 positive sera could induce typical CPE after 6–7 days characterized by cell aggregation, fusion, formation of vacuoles, and finally shedding. The other 19 positive sera inoculated C6/36 cells were consistent with mock cells (Fig. 1). The supernatants for the third passage were collected and sequenced to confirm the presence of DENV. A total of 25 isolates were positive for DENV nucleic acids, of which 20 showed CPE and 5 were no CPE. In these 25 isolated nucleic acids, 21 strains of DENV-1, 3 strains of DENV-2, and 1 strain of DENV-3 were identified according to the sequence of E protein. The 25 E proteins were sequenced, analyzed, and submitted to the GenBank database and are shown in Table 2.

The CPE (cytopathic effects) were observed in C6/36 cells infected with sera determined as flavivirus positive with flavivirus genus primer (6 days) (100 ×)
Summary of Information on Local Isolated Dengue Virus from the 2019 Outbreak in Xishuangbanna, Yunnan Province
Genetic and phylogenetic analysis of isolated DENV in Xishuangbanna
A phylogenetic tree was based on the nucleotide sequence of E gene of DENV which downloaded from NCBI database. Alignments of the selected sequences were performed by MegAlign in DNAStar software package with default parameters. Phylogenetic relatedness analyses of E gene nucleotide (DENV-1, DENV-2, DENV-4 1486 nt, DENV-3 1419 nt) were carried out using the MEGA6.1 program applying the neighbor-joining approach with 1000 bootstrap replicates. Following alignment and phylogenetic relatedness analyses, the E gene sequences of representing representative strains were selected to construct the phylogenetic tree.
E-gene genetic and phylogenetic analysis showed the information of isolated DENV in Xishuangbanna. According to the E gene sequences, 21, 3, and 1 of the 25 isolates were identified for DENV-1, DENV-2, and DENV-3, respectively (Fig. 2).
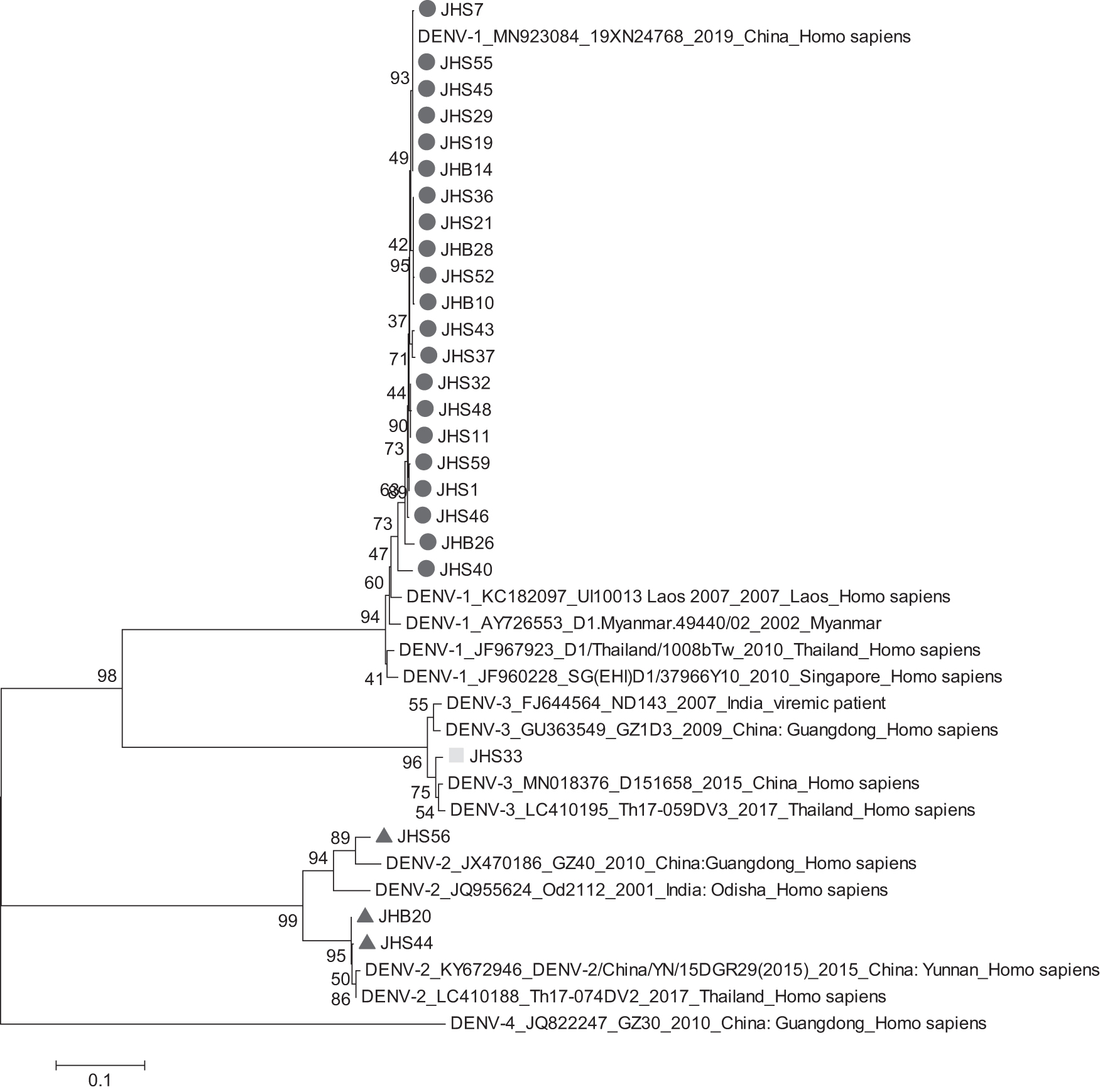
Phylogenetic relationship of E gene of isolated DENV with other DENV. DENV, Dengue virus.
Twenty-one isolated strains were located within the same branch as the DENV-1 strains prevalent in China, Thailand, Myanmar, Singapore, and Laos, with nucleotide homology ranging from 95.5% to 100% and amino acid homology ranging from 97.2% to 100% (Table 3). Figure 3 showed that the DENV-1 isolated strains in this study were Genotype I. Further analysis revealed that these 21 strains were located within 2 smaller branches, 20 of which belonged to the same cluster as the 2019 Guangdong epidemic strain and had the highest nucleotide and amino acid homology, while the other strain, JHS40, belonged to the same small branch as the 2010 Singapore strain and had the highest nucleotide and amino acid homology.

Phylogenetic relationship of E genes of isolated DENV-1 with other DENV-1.
Estimates of Evolutionary Divergence Over Sequence Pairs Within and Between Isolated DENV-1 Virus and Reference Strains, Based on Nucleotide (Up) and Amino Acid (Down)
The three isolated strains were located in the same branch as the DENV-2 from China, India, and Thailand in recent years, with nucleotide homology ranging from 90.4% to 99.5% and amino acid homology ranging from 96.2% to 99.7% (Table 4). Further analysis revealed that these three strains were located within two smaller branches, one branch was cosmopolitan genotype (JHS56) and another one was Asian-I genotype (JHB20 and JHS44). Further analysis showed that JHS56 was most closely related to the 2010 epidemic strain from Guangdong, China, while JHB20 and JHS44 were more closely related to the strains from Thailand and recent epidemics in Yunnan, China.
Estimates of Evolutionary Divergence Over Sequence Pairs Within and Between Isolated DENV-2 Virus and Reference Strains, Based on Nucleotide (Up) and Amino Acid (Down)
Virus strain JHS33 was located within the same branch as the DENV-3 strains from recent epidemics in China, Thailand, and India, with nucleotide homology ranging from 86.8% to 95.2%, and the amino acid homology ranged from 90.9% to 93.0% (Table 5).
Estimates of Evolutionary Divergence Over Sequence Pairs Within and Between Isolated DENV-3 Virus and Reference Strains, Based on Nucleotide (Up) and Amino Acid (Down)
Detection of unique mutations in the E genes of isolated DENV-1 in Xishuangbanna
To compare the genetic characteristics of isolated DENV-1 and other DENV-1 strains, reference DENV-1 in the same branch as shown in Fig. 4 was aligned and scanned using MEGAX, based on amino acid (Fig. 4). A higher degree of similarity was shown within those DENV-1 strains in the E gene. Relative to the reference DENV-1 strains, four unique amino acid mutations (D52, Y149, L312, T386) were discovered in the E gene of isolated DENV-1. D52 and Y149 were located at the domain II of DENV-1, while L312 and T386 were located at the domain III of DENV-1.

Schematic representation of DENV-1 E protein. Domain I is in white, domain II is in light gray, and domain III is in dark gray. A stem region (black) is embedded between the domain III and the transmembrane anchor (medium-gray). Unique mutations were shown in highlight.
The quaternary structure of the E in the prefusion conformation and postfusion conformation of JHS14 was predicted using the SWISS-MODEL online tool, based on the 6zqu or 3g7t structure (Fig. 5). The best model for the E dimer in the prefusion conformation had a high Seq-identity of 68.48%, a high GMQE-score of 0.79, and an estimated QMEANDisCo-score of 0.71 ± 0.05. The best model for the E trimer in the postfusion conformation had a high Seq-identity of 96.25%, a high GMQE-score of 0.64, and an estimated QMEANDisCo-score of 0.70 ± 0.05.
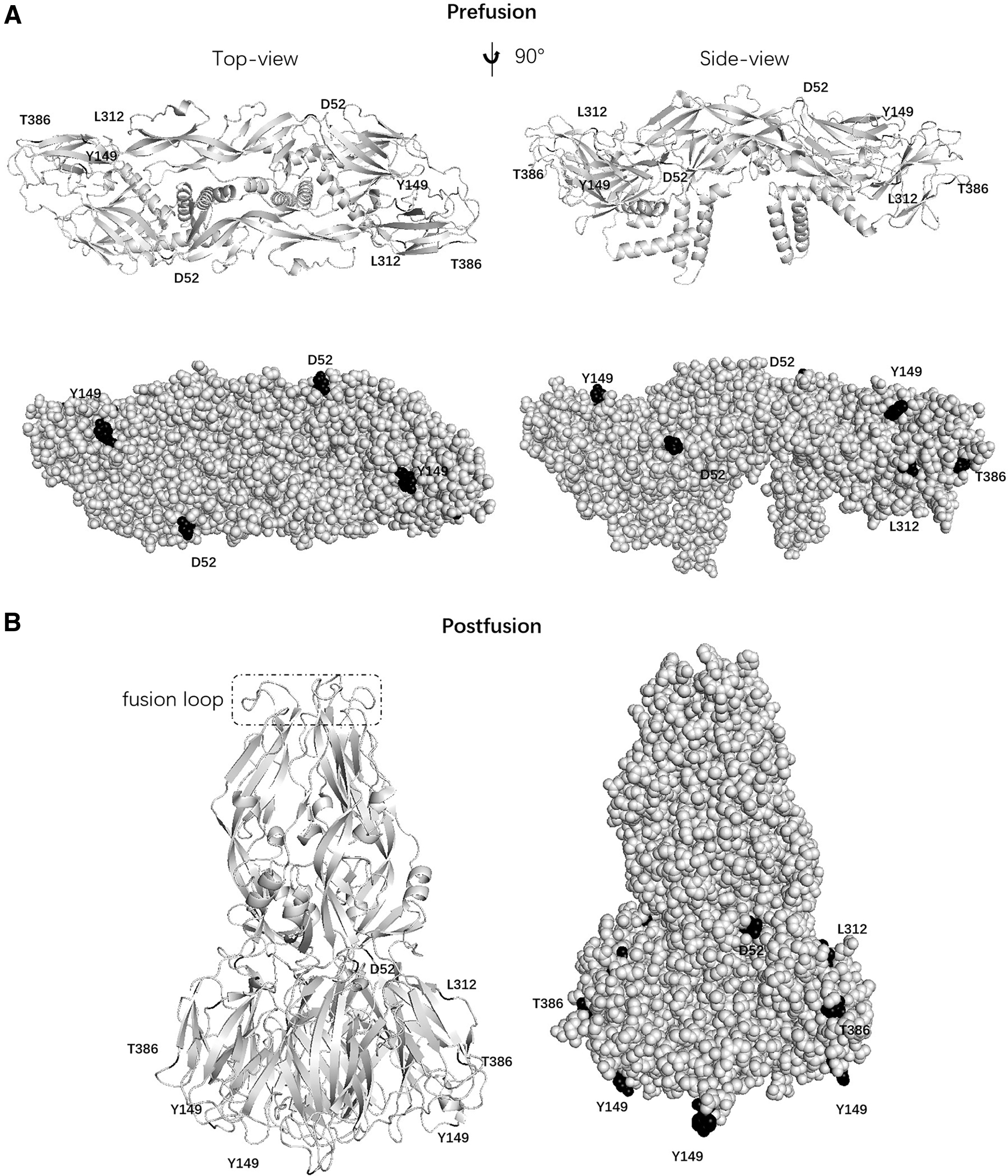
The predicated quaternary structure of E of JHS14.
Those scores indicated the correct topology of the two models. The two unique mutations (D52, Y149) in the E protein were predicted to be exposed on the protein surface in the prefusion conformation. All the four unique mutations were not predicted to form the fusion loop in the postfusion conformation.
Discussion
Yunnan is receiving increasing attention as a new epidemic site of DF that has emerged only in the last decade. Imported and local epidemics of the disease have occurred in the border areas of Yunnan Province between China and Myanmar and China and Laos for many years. Xishuangbanna is bordered by Laos and Myanmar to the east, west, and south and is adjacent to Thailand, Vietnam, and other Southeast Asian countries.
In 2013, 2015, and 2017, there were large-scale DF outbreaks caused by DENV-3, DENV-2, and DENV-1 in Xishuangbanna, respectively (Cao et al., 2019; Guo et al., 2015; JianHua et al., 2019). However, unlike the outbreak of single DENV serotype, it was three serotypes of DENV that should be responsible for the outbreak of DF in Xishuangbanna in 2019. In this study, although the 60 sera from patient were all NS1 antigen positive, only 39 sera were detected as DENV positive by specific primers. The disappearance of viremia could account for this phenomenon. A total of 25 DENV strains were isolated from patient sera, including 21 DENV-1 strains, 3 DENV-2 strains, and 1 DENV-3 strains. These abundant isolations of DENV-1 strains indicate that DENV-1 was the major serotype of the DF outbreak in Xishuangbanna in 2019.
According to the phylogenesis analysis of the E gene from the isolated DENV-1, the 22 DENV-1 strains belonged to the same cluster as the strain prevalent in Guangdong, China in 2019. Those results indicate that the DENV-1 strains responsible for the outbreak of DF in Guangdong and Xishuangbanna could have the same original. Notably, those strains of DENV were located within the same branch as the DENV epidemic in China, Laos, Thailand, and Vietnam in recent years, indicating that DENV was transmitted by the DENV-infected patient from neighboring countries and local mosquitoes, leading to a local epidemic in Xishuangbanna in 2019.
The E of isolated DENV-1 contains four novel unique mutations (D52, Y149, L312, T386) that have never been reported before. The E protein is a structural protein located on the surface of DENV and consists of 495 amino acids (Venkatachalam and Subramaniyan, 2014). E protein has three main domains, including domain I, domain II, and domain III (Eerde et al., 2019). We identified two unique mutations in each of structural domains II and III, when comparing isolated DENV-1 with other reference DENV-1. Two unique mutation sites (D52, Y149) are exposed on the surface of the predicated quaternary structure of the E dimer (prefusion conformation) of JHS14, indicating that D52 and Y149 might affect the binding affinity between E protein and its receptors.
Structural domain II causes a conformational change to form the postfusion conformation upon pH change, allowing structural domain III to mediate the fusion of the virus with the host receptor (Park et al., 2020). Although some residues in E protein did not make up the fusion loop, a set of residues was critical for fusion loop functionality. H149 has been reported as a switch triggering the exposure of fusion loop (Christian et al., 2013). Thus, Y149 might affect fusion loop exposure. In addition, structural domain III contains type- and subtype-specific antigenic epitopes that induce neutralizing antibodies (Hu et al., 2019). Many studies have reported that V312 in domain III of E in DENV was a key amino acid residue of antibody binding protein. Hence, L312 mutations might escape from the neutralizing antibodies (Lisova et al., 2007; Thullier et al., 2001; Thullier et al., 1999). Thus, the role of those four unique mutations of E of isolated DENV-1 in the entry of virus and neutralizing activity needs further analysis.
In conclusion, this study has reported the characteristics of DENV causing DF outbreaks in Xishuangbanna in 2019, which can provide a reference for the future analysis of DENV in virulence, replication, infection, pathogenicity, and vaccine. Xishuangbanna is connected to Laos and Myanmar in the southeast, south, and southwest, respectively, and is adjacent to Thailand and Vietnam, with frequent human traffic, which greatly increases the possibility of DENV being imported from different sources and causing local epidemics. Therefore, it is urgent to strengthen the surveillance of DENV in border areas, discover the source of the virus, and take effective preventive and control measures.
Footnotes
Ethics Approval and Consent to Participate
The experimental protocol was established, according to the legal and ethical approvals by the Ethics Review Committee of Yunnan Animal Science and Veterinary Institute. The experiments were performed in accordance with the relevant guidelines and regulations.
Research Involving Humans
All clinical investigations should be conducted according to the Declaration of Helsinki principles.
Author Disclosure Statement
No conflicting financial interests exist.
Funding Information
This study was funded by Yunnan Chenggong expert workstation (202005AF150034); Basic Research Projects of Yunnan Province (202301AT070028, 202201AS070062); Projects funded by the central government to guide local scientific and Technological Development (202207AB110006); Science and Technology Innovation Team Construction Project of Kunming Medical University (CXTD202111).