
Abstract
Background:
Haemagogus janthinomys is a primary sylvan vector of yellow fever virus and the emerging Mayaro virus. However, despite its medical importance, there is a dearth of data on the molecular taxonomy of this mosquito species.
Methods:
In this study, DNA barcoding analysis was performed on 64 adult female mosquitoes from Trinidad morphologically identified as Hg. janthinomys. The mitochondrial cytochrome c oxidase I (COI) gene and ribosomal DNA internal transcribed spacer 2 (ITS2) region of the mosquitoes were PCR amplified and sequenced, and molecular phylogenies inferred.
Results:
The BLASTN analysis showed that only 20% (n = 13/66) of COI sequences had high similarity (>99% identity) to Hg. janthinomys and the remaining sequences had low similarity (<90% identity) to reference GenBank sequences. Phylogenetic analysis of COI sequences revealed the presence of four strongly supported groups, with one distinct clade that did not align with any reference sequences. Corresponding ITS2 sequences for samples in this distinct COI group clustered into three clades.
Conclusions:
These molecular findings suggest the existence of a putative new Haemagogus mosquito species and underscore the need for further, more in-depth investigations into the taxonomy and classification of the Haemagogus genus.
Introduction
Haemagogus mosquito species are the main vectors associated with maintaining the enzootic cycle of yellow fever virus and the emerging Mayaro virus (Hoch et al., 1981; Izurieta et al., 2018; Mota et al., 2015; Pego et al., 2014). The Haemagogus janthinomys and Haemagogus leucocelaenus mosquitoes are the two most important species of epidemiological significance since they have been linked to recurrent isolations of these two viruses (De Abreu et al., 2019; Long et al., 2011). These canopy dwelling mosquitoes of South and Central America are primarily found in tropical rain forests where they breed mainly in bamboo stumps and tree holes (Alencar et al., 2018; Arnell, 1973). They were found in Trinidad and Tobago, and in the small islands off Trinidad, and Jamaica (Ali et al., 2019; Chadee and Tikasingh, 1991; Chadee and Tikasingh, 1989).
Mosquitoes belonging to the Haemagogus genus include 28 species within 2 subgenera: Haemagogus (24 species) and Conopostegus (4 species); they are well known for their characteristic shiny metallic coloration and show peak activity during midday hours (Arnell, 1973; Chadee and Tikasingh, 1991). An earlier report documented Haemagogus celeste, Haemagogus equinus, Hg. Janthinomys, and Hg. leucocelaenus as the four main species present in Trinidad (Arnell, 1973; Zavortink, 1972).
Identification of mosquito species plays an important role in vector-borne disease risk assessment and in the development of control strategies in case of any possible arboviral outbreaks. Traditional morphology-based approaches have been the most common method for taxonomic identification of mosquitoes, including Haemagogus species. Several earlier studies in Trinidad have applied these traditional techniques to show the presence of Haemagogus mosquitoes, mainly in the northwestern peninsula of the island (Chadee and Tikasingh, 1991; Chadee and Tikasingh, 1989; Chadee et al., 1995; Chadee et al., 1993; Chadee et al., 1992). More recently, Ali et al. (2019) also used these classical methods in an updated island-wide survey that showed broad distribution of Haemagogus mosquitoes throughout the island.
Despite their popularity, morphological methods are unable to distinguish among members of mosquitoes that form cryptic species or species complexes, since they may have similar physical features but may be genetically and behaviorally divergent (Lobo et al., 2015). Distinguishing among species in these groups is important since different genotypes may have variable behavioral and functional traits such as host feeding preferences, pathogen competence, and insecticide resistance. Hence, alternative molecular-based methods need to be developed to differentiate among these related mosquitoes. DNA barcoding is a more recent approach that uses PCR amplification and sequencing of targeted molecular markers such as the mitochondrial cytochrome c oxidase I (COI) and ribosomal DNA internal transcribed spacer 2 (ITS2) to classify and identify a wide range of mosquito species based on their unique genetic signatures (Beebe, 2018).
This strategy, in combination with traditional morphological methods, has gained popularity in vector surveillance, taxonomy, and genetic diversity studies (Ashfaq et al., 2014; Weeraratne et al., 2017). However, molecular-based methods have not yet been applied extensively to genetically characterize Haemagogus mosquitoes and there were no such previous attempts in the Caribbean region. In the current study, the traditional taxonomic keys and DNA barcoding technique using the mitochondrial COI gene and the chromosomal ITS2 region were used to characterize Hg. janthinomys mosquitoes collected from forested locations on the island of Trinidad. The goal of the study was to compare methods and determine if DNA barcoding can be used as a suitable tool for accurate identification of Hg. janthinomys.
Materials and Methods
Mosquito collection and identification
Hg. janthinomys adult females were collected using human bait landing catches (Chadee et al., 1992) from 12 forested sites (Fig. 1) in 2018, 2019, and 2020. The collected specimens were identified and recorded using the published taxonomic keys (Arnell, 1973; Darsie, 1985; WRBU, 2012). A secondary confirmation of the identity of the mosquitoes was also done by geometric morphometric wing analysis using 14 landmarks based on natural anatomical junctions of the veins, as described by Silva et al. (2019).
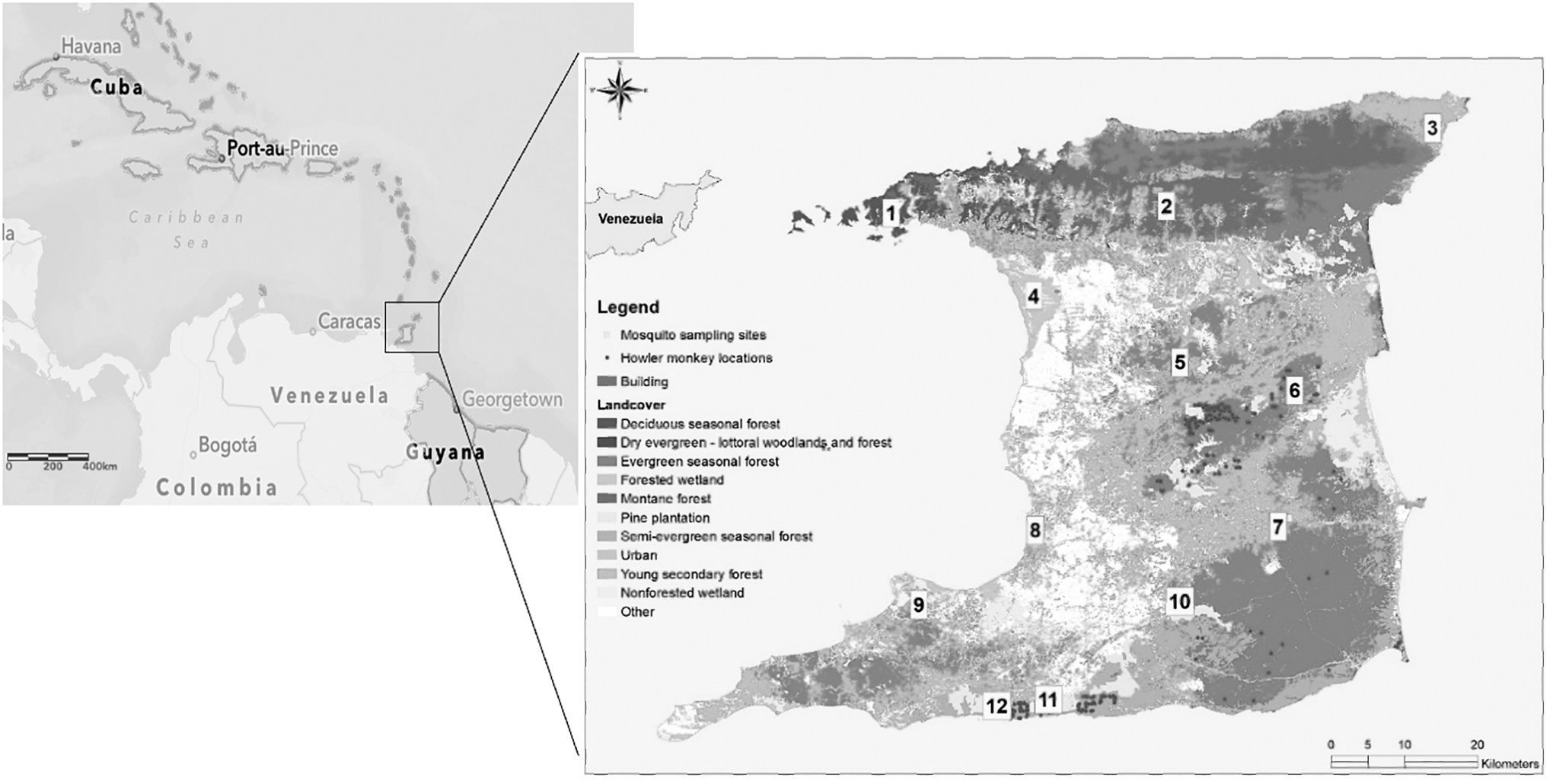
Trinidad Haemagogus janthinomys mosquito sampling sites in 12 forested locations 1: Chaguaramas, 2: Hollis, 3: Cumana, 4: Caroni Swamp, 5: Mamoral, 6: Mt. Tamana, 7: Ecclesville, 8: Claxton Bay, 9: Rousillac, 10: Catshill, 11: Morne Diablo, 12: Quinam. Adapted from Ali et al. (2019).
DNA extraction and barcoding
The genomic DNA from individual insect specimens (n = 2–9, from each location), as well as from a laboratory colony (Parasitology Laboratory, UWI, St. Augustine Campus) established from individual specimens collected at the Caroni swamp location, was extracted using the DNasy Blood & Tissue Kit (Qiagen) according to the manufacturer's instructions. DNA was also extracted from the three ethanol-preserved Haemagogus mosquito samples that had been collected and morphologically identified in 2006 by the Insect Vector Control Division (IVCD) of the Ministry of Health, Trinidad and Tobago, using a rapid standard alkaline-lysis protocol (Rudbeck and Dissing, 1998). PCR amplification (25 μL reaction volume) of the COI barcoding gene was done using the primers LCO1490 (5′-GGTCAACAAATCATAAAGATATTGG-3′) and HCO2198 (5′-TAAACTTCAGGGTGACCAAAAAATCA-3′) (Ivanova et al., 2007; Vrijenhoek, 1994); ITS2 barcoding was done using primers ITS2A (5′-TGTGAACTGCAGGACACAT-3′) and ITS2B (5′-TATGCTTAAATTCAGGGGGT-3′) (Ashfaq et al., 2014; Beebe et al., 2007).
The PCR cycling conditions for COI were as follows: 1 cycle at 94°C for 2 min; 40 cycles at 94°C for 30 s, 49°C for 45 s, and 72°C for 45 s; and a final extension step at 72°C for 10 min. PCR cycling conditions for ITS2 were as follows: 1 cycle at 94°C for 5 min; 35 cycles at 94°C for 1 min, 55°C for 1 min, and 72°C for 2 min; and a final extension step at 72°C for 5 min.
The PCR products were run on a 1% agarose gel and the discrete positive bands were purified by the Wizard SV gel and PCR clean up kit (Promega). Sanger sequencing (COI and ITS2) of the purified products was performed on the 3730xl DNA analyzer (Applied Biosystems) at the Genomic and Bioinformatics Core facility (
Phylogenetic analysis
Clustal W implemented in MEGA X 10.0.5 software (Kumar et al., 2018) was used to align the generated nucleotide sequences as well as reference COI from Haemagogus mosquito species available from GenBank. However, there were no Hg. celeste COI sequences available in GenBank. The aligned sequences were exported in nexus format and the base pair substitution model of the aligned sequence matrix was determined using the jModelTest (v2.1.10) program (Posada, 2008). Phylogenetic trees for COI and ITS2 sequences were then searched under the Akaike information criterion and the Bayesian information criterion of maximum likelihood in the MEGA X 10.0.5 software (Kumar et al., 2018), with bootstrap set at 1000 pseudoreplicates.
Species delimitation
Delimitation was performed for the COI sequences using the Assemble Species by Automatic Partitioning (ASAP) webserver (
Data availability
The data that support the findings of this study are openly available in the NCBI GenBank database (
Results
Adult females (n = 63) collected from all the sample sites and the laboratory colony strain were identified as Hg. janthinomys mosquito species using the morphological-based taxonomic keys (Arnell, 1973; Darsie, 1985; WRBU, 2012). The wing geometric morphometric analysis also confirmed that the mosquitoes had features typical of Hg. janthinomys and not those of the closely related Haemagogus capricornii as described by Silva et al. (2019). Adult male mosquitoes were also collected, and none was identified as Hg. capricornii. Furthermore, the mosquitoes (n = 3) collected by the IVCD during 2006 had, at that time, been morphologically identified as Hg. janthinomys and Hg. leucocelaenus.
The BLASTN analysis of the COI gene sequences from all mosquitoes included in this study revealed that 73% (n = 48/66) had low similarity scores (<90%) to sequences in NCBI's GenBank database. However, 13 sequences from mosquitoes morphologically identified as Hg. janthinomys from Point Gourde, Chaguaramas (n = 2), Catshill (n = 5), Hollis (n = 5), and Caroni (n = 1) had high similarity scores (>99%) to Hg. janthinomys reference sequences. The phylogenetic analysis of the COI target resulted in sequences grouping into four strongly supported clades in the maximum likelihood tree (Fig. 2). The 13 sequences that had high similarity to Hg. janthinomys all grouped together in the Hg. janthinomys clade, with reference sequences of this species originating from French Guiana, Brazil, and Colombia. Sequences from other specimens clustered strongly into clades with reference sequences from Hg. equinus and Hg. leucocelaenus.

Maximum likelihood phylogenetic tree of COI and GenBank sequences showing clades corresponding to the molecular identification of Haemagogus mosquitoes collected in Trinidad. The Akaike information criterion (TIM2 + 1+G) model tree is represented above. Both the Akaike information criterion and the Bayesian information criterion models resulted in similar tree outputs. COI, cytochrome c oxidase I.
Phylogenetic analysis revealed a clade that does not correspond closely to any Haemagogus reference sequence and may represent a putative new species. Specimens clustered in this clade were geographically the most widely distributed, originating from all sampling locations except Catshill and Hollis (Table 1). This clade also contained sequences of two mosquitoes collected in 2006 that were morphologically identified as Hg. leucocelaenus. The intraclade diversity for COI sequences of the new Hg. species was 0.03%; while interspecies diversity between clades was found to be 0.1%. The pairwise genetic distance matrix (Table 2) also indicated that Trinidad Hg. janthinomys, Hg. Equinus, and Hg. leucocelaenus had greater genetic distances (0.138–0.157) to the putative new species than distances among themselves (0.080–0.119).
Frequencies of Trinidad Haemagogus Mosquito Species Identified from Different Collection Sites
Pairwise Genetic Distances Between Representative Trinidad Haemagogus Mosquito Species
GenBank accession numbers.
Phylogenetic analysis of ITS2 sequences from specimens in the unidentified Haemagogus COI clade clustered into three well-supported ITS2 clades, with the two Pt. Gourde sequences forming a distinct group (Fig. 3). The intraspecies diversity within this clade was found to be 0.07%. Sequence alignments showed several regions of hypervariability in the COI region (Fig. 4), where relatively more substitutions were observed in the putative new Haemagogus species. The best scores of ASAP partitioned species into four groups for both COI- and ITS2-based sequences, with specimens falling into similar groupings as observed for phylogenetic trees (putative species, Hg. equinus, Hg. Leucocelaenus, and Hg. janthinomys). The resulting match ratio was 2.50 for COI and 1.50 for ITS partitioning.
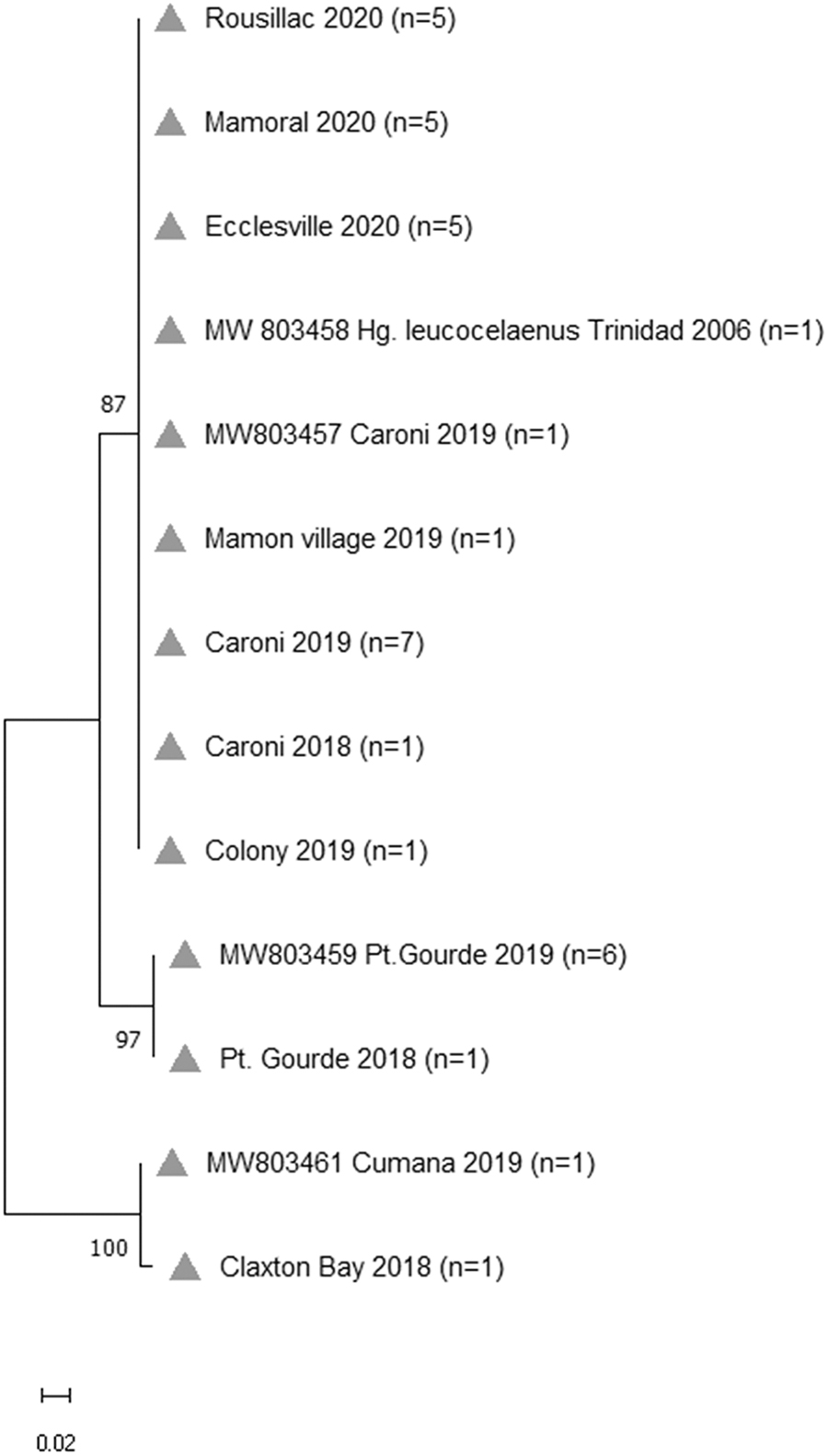
Maximum likelihood phylogenetic tree of putative new Haemagogus species ITS2 sequences corresponding to the molecular identification of Haemagogus mosquitoes collected in Trinidad. The Akaike information criterion (TIM2 + 1+G) model tree is represented above. Both the Akaike information criterion and the Bayesian information criterion (k80) models resulted in similar tree outputs. ITS2, internal transcribed spacer 2.
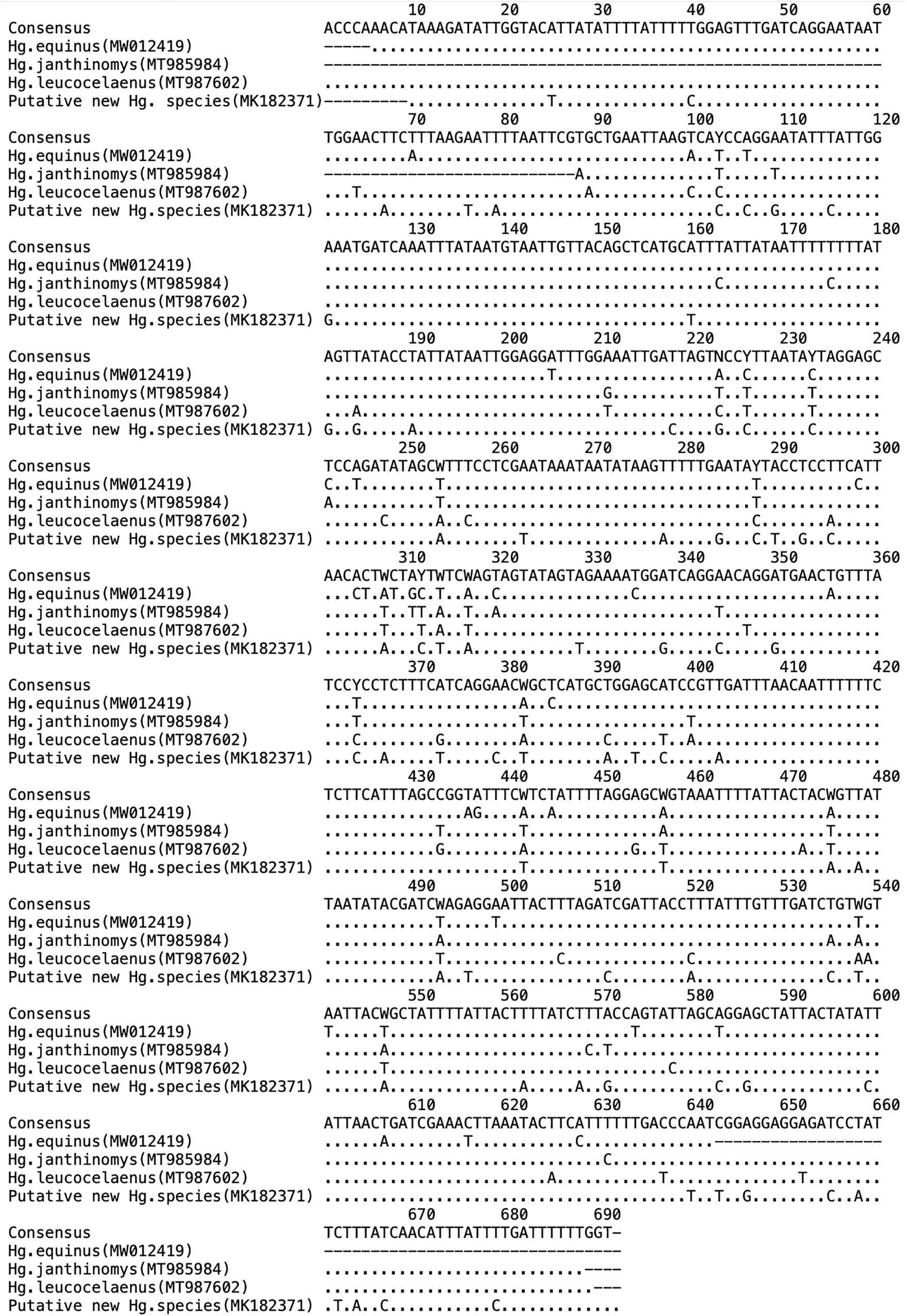
Nucleotide substitutions at one variable region of the COI gene of Haemagogus mosquitoes.
Discussion
Advances in molecular techniques have demonstrated their value as a surveillance tool for vector identification (Beebe, 2018; Hoffman et al., 2021; Kirchgatter et al., 2023; Zhong et al., 2020), and have also allowed the identification of cryptic species based on genetic differences among morphologically identical mosquitoes (Hoffman et al., 2021; Lobo et al., 2015; Minard et al., 2017). The current study has shown evidence of a possible undescribed Haemagogus species with identical morphology to Hg. janthinomys. Of the 64 adult females morphologically identified as Hg. janthinomys (Arnell, 1973; Darsie, 1985; WRBU, 2012), only 13 specimens had COI gene sequences that were highly similar (>99% identity) to the published Hg. janthinomys reference sequences, while the vast majority had a low similarity (<90% identity) to any reference sequences in the NCBI GenBank.
The large disparity in similarity levels between the COI sequences of the local mosquitoes is suggestive of the occurrence of a previously unrecognized Haemagogus mosquito species. Phylogenetic analysis also confirmed the existence of a distinct genotype, clustering into a separate clade, which accounted for most samples, and was the most widely distributed clade on the island. Likewise, the data provide evidence of a putative new Haemagogus mosquito species in Trinidad, which possess indistinguishable morphological features to that of Hg. janthinomys. High similarity in morphology between Haemagogus genotypes has already been reported by Silva et al. (2019) who were unable to separate Hg. janthinomys from Hg. capricornii using standard taxonomic keys, but the application of an alternative geometric morphometry method based on wing structures was able to distinguish between the two species. However, this alternative technique failed to distinguish among the Hg. janthinomys genotypes from Trinidad.
The occurrence of cryptic species has been reported in several Culicidae species, including the Aedes albopictus subgroup (Minard et al., 2017), Culex pipiens (Dumas et al., 2016), and is common in the genus Anopheles (Hoffman et al., 2021; Zhong et al., 2020). Earlier studies that identified cryptic mosquito species were based on investigating functional traits such as vector competence for pathogens. For example, competence for the malaria parasite by Anopheles mosquitoes, which also had important epidemiological implications. The use of DNA-based technologies such as DNA barcoding makes it much easier to distinguish among morphologically identical mosquitoes. Thus, further investigations are needed on Haemagogus mosquitoes to determine if there is a genetic basis for behavioral patterns, as has been demonstrated for other mosquito species.
The differentiation between vector and nonvector in mosquitoes is important for control, since populations could be comprising dominant mosquito genotypes that may be competent in disease transmission and/or may be insecticide resistant (Bickford et al., 2007; Zheng, 2020).
The ITS2 region may have good potential for use as a main DNA barcoding target in the identification of morphologically indistinguishable species of Haemagogus mosquitoes. The potential of the chromosomal ITS2 region in barcoding-based identification of mosquitoes such as Anopheles and Aedes has been well documented (Beebe, 2018; Fang et al., 2017; Minard et al., 2017). The presence of nucleotide substitutions and indels in the hypervariable region of the ITS2 of the Haemagogus mosquitoes may also offer an opportunity for developing rapid multiplexed PCR-based diagnostic systems for efficient discrimination of species or subspecies without the need for DNA sequencing.
It should be noted that during exploration of GenBank reference COI sequences of other Haemagogus species, discrepancies for Hg. leucocelaenus from South America were also discovered, in that they did not cluster with other Hg. leucocelaenus sequences. This strongly suggests that previous morphological identifications of adult Haemagogus species in Trinidad and the Neotropics may be presumptive and may need verification through molecular-based identification methods such as genetic barcoding. Although these results show a relatively low prevalence of Hg. janthinomys that are closely related to those in South America, the unique genotype may be a result of divergence occurring after Trinidad separated from the South American continent. However, it is also possible that the putative new species may be present on the mainland but are yet to be identified due to the limited genetic studies of Haemagogus mosquitoes.
Furthermore, this study also suggests the need for investigating the competence of the new putative species as a potential vector for arboviral disease due to its widespread distribution and proximity to urban areas throughout the island.
Conclusions
This study highlights the importance of taxonomic identification and molecular identification as complementary techniques that can be used together to obtain a more accurate identification of mosquitoes, especially for cryptic species. The findings confirm the need for additional molecular investigations on the medically important Haemagogus mosquito species across the region to acquire representative COI and ITS2 reference sequences for better understanding of the vector's population dynamics. Accurate species identification using these techniques can bridge gaps in mosquito taxonomy that would contribute to a better understanding of vector populations.
Footnotes
Acknowledgments
The authors would like to thank Rachel Shui Feng, Lester D. James, Nikhella Winter, Christopher de Caires-Glenn, and members of the Insect Vector Control Division, Ministry of Health, and Trinidad & Tobago for assistance in the field.
Authors' Contributions
R.A. and R.D.L. collected and morphologically identified mosquitoes; R.A. performed nucleic acid extraction; R.A. and A.R. undertook analysis and wrote the article with input from all the authors; D.D.L. and J.M.C. sequenced and submitted representative samples to the GenBank database. A.M., J.J., C.V.F.C., B.D., and D.W.S. provided advise, logistic support, and wrote the article with all the authors.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.