
Abstract
Background:
Aedes aegypti, is the primary vector of dengue, Chikungunya, Zika, and yellow fever viruses. Both natural and human-impacted landscapes have selective pressures on Ae. aegypti, resulting in strong genomic structure even within close geographical distances.
Materials and Methods:
We assess the genetic structure of this medically important mosquito species at the northern leading edge of their distribution in Southwestern USA. Ae. aegypti were collected during 2017 in the urban communities of El Paso and Sparks, Texas (USA) and in the city of Ciudad Juárez, Mexico.
Results:
Thousands of nuclear loci were sequenced across 260 captured Ae. aegypti. First, we recovered the genetic structure of Ae. aegypti following geography, with all four major collection communities being genetically distinct. Importantly, we found population structure and genetic diversity that suggest rapid expansion through active-short distance dispersals, with Anapra being the likely source for the others. Next, tests of selection recovered eight functional genes across six outliers: calmodulin with olfactory receptor function; the protein superfamily C-type lectin with function in mosquito immune system and development; and TATA box binding protein with function in gene regulation.
Conclusion:
Despite these populations being documented in the early 2000s, we find that selective pressures on specific genes have already occurred and likely facilitate Ae. aegypti range expansion.
Introduction
Aedes aegypti is the primary global vector of dengue, Chikungunya, Zika, and yellow fever virus, (Bogoch et al., 2016; Kang et al., 2018; Weaver & Lecuit, 2015). Natively found in tropical climates of Africa, their close association to and successful exploitations of human-made environments has made Ae. aegypti a highly successful invasive species with a global distribution across six continents (Cosme et al., 2020; Schmidt et al., 2021). Their range expansion appears to be based on long- and short-range passive and active dispersal (Hengeveld, 1989; Schmidt et al., 2020), resulting in population establishment via serial founder events (Brown et al., 2011; Brady and Hay, 2020; Pless et al., 2022).
Advancements in next-generation sequencing methodology and decreasing costs provide opportunities to assay thousands of nuclear markers over landscape-level sampling efforts to understand population interconnectedness and resulting range expansions (Davey et al., 2011; B Li et al., 2018; Schmidt et al., 2021). In fact, partial-genome sequencing resulting in thousands of nuclear single nucleotide polymorphisms (SNPs) has helped shed light on the population structure of Ae. aegypti worldwide: Brazil (Ayres et al., 2003); Venezuela (Herrera et al., 2006); Puerto Rico (Apostol et al., 1996), and Mexico (Gorrochotegui-Escalante et al., 2002). Together, these studies have found that spatial distance, including urbanization levels and resource availability, are primary influencers of Ae. Aegypti population structure (Maffey, et al. 2020; Maffey, et al., 2022); with short distance dispersal (Bosio et al., 2005; Costa-Ribeiro et al., 2006; Julio et al., 2009; Ocampo & Wesson, 2004; Paupy et al., 2004), including between proximate neighborhoods (Gloria-Soria et al., 2016; Herrera et al., 2006) resulting in unique population structure in Ae. aegypti. As a result, coupling partial-genome sequence data with landscape-level sampling can help determine the local genetic structure of Ae. aegypti populations (Davey et al., 2011; B Li et al.).
Here, we attempt to better understand the invasive mosquito, Ae. aegypti, in the temperate climate of southwestern United States and Northern Mexico. To do so, we assessed the genetic structure across two unincorporated urban communities located along the U.S.–Mexico border that represent Ae. aegypti most northward range limit today (Gloria-Soria et al., 2016; Lozano-Fuentes et al., 2009). Given that Ae. aegypti were identified in the El Paso community as early as 2003 (Merrill et al., 2005), we hypothesize sampled Ae. aegypti will show fine-scale genetic structure even between geographically close locations through biological processes (Hopperstad et al., 2021; Pless et al., 2022). If we recover population structure, we will test for signatures of selection between genetically defined groups. Despite the relative recency of their invasion (Merrill et al., 2005), short generation time of approximately 10–15 per year (Gloria-Soria et al., 2016; Crawford, et al., 2017), and relatively high fecundity with average clutch size of 100 eggs (Joy, et al., 2010), Ae. aegypti may result in apparent selective pressures from local ecologies to be evident. As a result, we expect to find a handful of loci linked to selectively important genes under local selection (Gaburro et al., 2018). In the end, we aim to elucidate invasion processes and the potential risk of arboviruses associated with Ae. aegypti by understanding genetic structure (Evans et al., 2015; Soghigian et al., 2020); which together, can facilitate the development of targeted vector control measures to limit future Ae. aegypti dispersal (Burford Reiskind et al., 2016; Rose et al., 2020).
Materials and Methods
Sampling, identification, and DNA isolation
Adult Ae. aegypti mosquitoes were collected from June to December 2017 in two urban unincorporated communities located along the U.S.–Mexico border: (1) community of Sparks located in El Paso County, Texas, (2) two additional neighborhoods within the city of El Paso, Texas, and (3) community of Anapra found in Ciudad Juarez, Mexico (Fig. 1). Gravid traps placed inside and outside houses were used to capture mosquitoes (BioQuip Products, Inc., California). All adult mosquitoes were identified morphologically based on a dichotomous key developed by Darsie and Ward (2005). The male Ae. aegypti was placed individually in 2.0 mL cryogenic tubes, whereas the identified female was first dissected, with head, legs, and wings placed in 2.0 mL cryogenic tubes and frozen at −80°C. Total DNA was extracted across a total of 265 mosquitoes using the Promega Wizard Genomic DNA Purification Kit, following the manufacturer’s protocols (Promega, Madison, WI). DNA quality was based on the presence of a high molecular weight band, visualized with 1% agarose gel electrophoresis and quantified using a Qubit 3 Flourometer (Invitrogen, Carlsbad, CA) with a minimum concentration of 20 ng/µL.

Location of captured Aedes aegypti in 2017 in Sparks, Anapra, and two locations found in different regions of the City of El Paso that are separated by the Franklin Mountains.
We followed procedures described by Lavretsky et al., (2015) to create double digest restriction-site associated DNA (ddRAD-seq) libraries, with double-sided magnetic bead-based fragment size selection protocols following Hernández et al., (2021). We enzymatically fragmented genomic DNA using SbfI and EcoRI restriction enzymes, and ligated Illumina TruSeq compatible barcodes and permitted future de-multiplexing. All libraries were pooled in equimolar concentrations, and 150 base pair (bp), single-end (SE) sequencing was completed on an Illumina HiSeq X at Novogenetics Ltd. (Sacramento, CA). Illumina reads were deposited in NCBI’s Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra; accessed on 27 March 2024; BioProject PRJNA1092587; Bio Sample Accession Numbers SAMN40627824–SAMN40628083).
Raw-illumine reads were demultiplexed using the ddRADparser.py script of the BU ddRAD-seq pipeline (DaCosta and Sorenson 2014) based on perfect barcode/index matches. Next, custom in-house Python scripts (Python scripts available at https://github.com/jonmohl/PopGen; see Lavretsky et al., 2020) were used to automate sequence filtering, alignment, and genotyping using a combination of Trimmomatic (Bolger et al., 2014), Burrows Wheeler Aligner v. 07.15 (BWA; Li & Durbin, 2009), and SAMTOOLS v. 1.7 (Bolger et al., 2014). We used VCFtools v. 0.1.15 (Danecek et al., 2011) to filter VCF files for any bp missing >10% of samples that also included a minimum bp depth of 5× (i.e., 10× per genotype) and quality per base PHRED scores of ≥30. All genomic data was aligned to Ae. aegypti genome AaegL5 (Matthews et al., 2018).
Genetic structure and molecular diversity of Ae. aegypti
Before analyses, we used PLINK v. 1.9 (Purcell et al., 2007) to ensure that singletons (i.e., minimum allele frequency [maf] = 0.0038) and any SNP missing >10% of data across samples were excluded in each dataset. In addition, identified independent SNPs by conducting pair-wise linkage disequilibrium (LD) tests across ddRAD-seq autosomal SNPs (–indep-pairwise 2 1 0.5) in which one of two linked SNPs are randomly excluded if an LD correlation factor (r 2) > 0.5 was obtained. We conducted all analyses without a priori information on location or population identity.
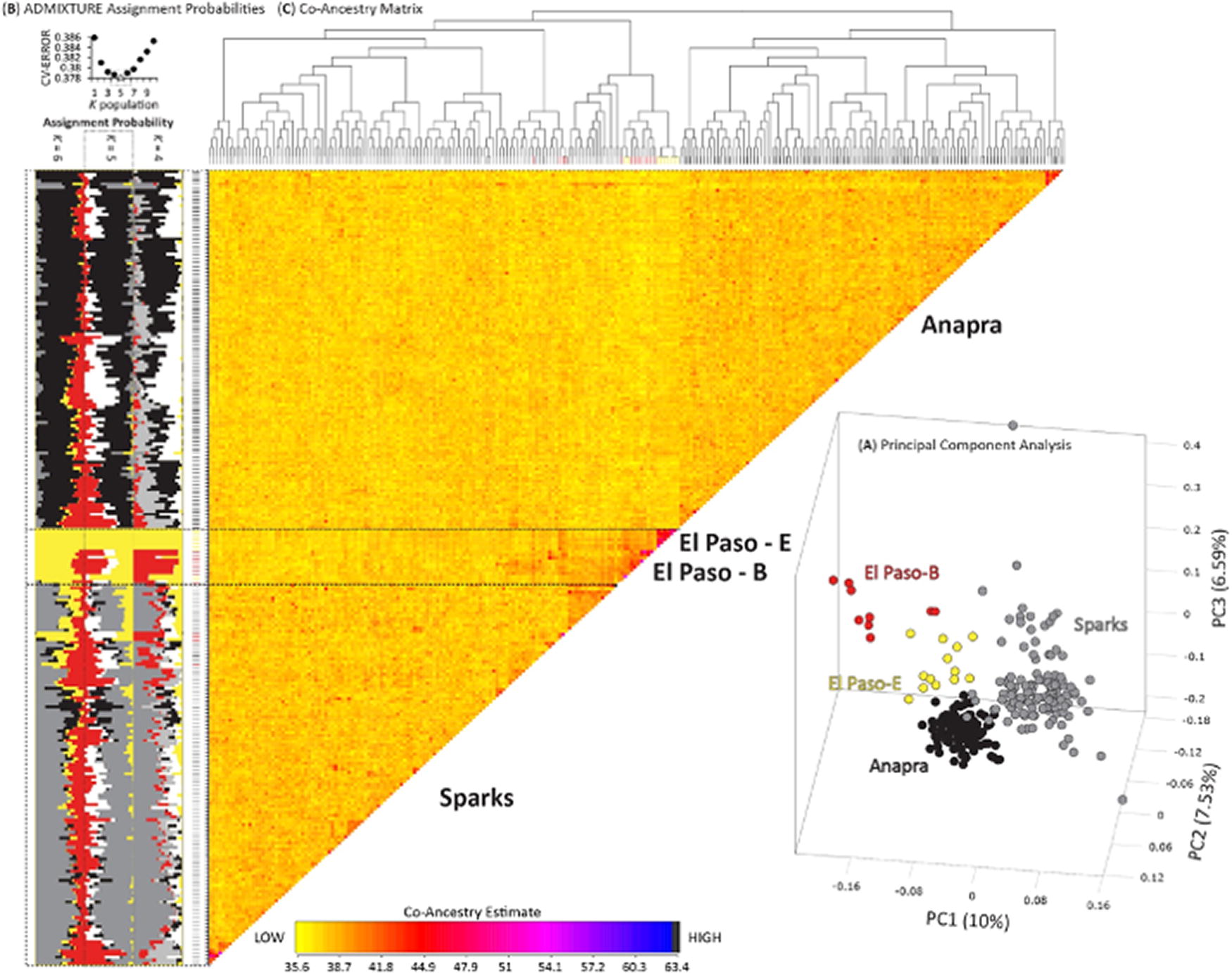
Genetic structure was based on an independent biallelic set of ddRAD-seq SNPs. First, a principal component analysis (PCA) was conducted using the dudi.pca function in R (Dray & Dufour, 2007; Jombart et al., 2008). Next, individual assignment probability estimates were derived from the program ADMIXTURE v.1.3 (Alexander et al., 2012; Alexander & Lange, 2011). ADMIXTURE analyses were run for K population models 1–10, with a 10-fold cross-validation, and with a quasi-Newton algorithm employed to accelerate convergence (Zhou et al., 2011). Each analysis used a block relaxation algorithm for point estimation and terminated once the change in the log-likelihood of the point estimations increased by <0.0001. Each K population was evaluated 100 times, with the optimum K population model based on the lowest average CV-error score across the 100 analyses for each K. The R package PopHelper (Francis, 2016) was used to convert ADMIXTURE outputs into CLUMPP input files at each K value. The robustness of the individual assignments to genetic clusters at each K value with the program CLUMPP version 1.1 (Jakobsson and Rosenberg 2007). In CLUMPP, we employed the Large Greedy algorithm and 1,000 random permutations. Final ADMIXTURE proportions for each K value and per sample assignment probabilities (Q estimates; the log-likelihood of group assignment) were based on CLUMPP analyses of all 100 replicates per K value. Finally, we evaluated patterns of coancestry using fineRADstructure (Malinsky et al., 2018) that infers a matrix of coancestry coefficient based on the distribution of identical or nearest neighbor haplotypes among samples. A burn-in of 100,000 iterations, followed by 100,000 Markov chain Monte Carlo iterations were completed, followed by tree building using default parameters. To visualize the results, we used the R scripts fineradstructureplot.r and finestructurelibrary.r (available at http://cichlid.gurdon.cam.ac.uk/fineRADstructure.html).
We used VCFtools v. 0.1.15 (Danecek et al., 2011) for per population nucleotide diversity (π; — site-pi), as well as estimated pairwise population relative divergence (FST; — weir-fst-pop) across loci. Note that these analyses included all possible variant and nonvariant sites that met filtering criteria.
Outlier analyses
Statistical outliers were evaluated using the BayeScan v.2.1 (Foll & Gaggiotti, 2008) program, which has relatively low rates of false positives (<1%) for populations with low overall differentiation (Pérez‐Figueroa et al., 2010). BayeScan employs a reversible-jump MCMC method by calculating a posteriori probability model with and without selection across loci. The program also distinguishes between positive/diversifying selection (α > 0) and balancing/purifying selection (α < 0). Analyses included 20 pilot runs of 5,000 steps each, followed by 100,000 burn-in steps and 10,000 sampling steps with a thinning interval of 10 for a total of 200,000 iterations. The prior odds parameter for the neutral model was set at log10(10) (Posterior Odds > 1.0), and we used a false discovery probability of 0.05. Finally, genes that may be linked to any locus found to be potentially under directional selection were evaluated by searching 10,000 bps upstream and downstream; which is within a linkage group of Ae. aegypti (Matthews et al., 2018).
Results
We successfully sequenced ddRAD-seq libraries for 97.6% (122/125), 98.3% (116/118), and 100% (22/22) of mosquitoes from Sparks, Anapra, and El Paso, respectively. A total of 40,123 aligned bps across 1,859 ddRAD-seq autosomal loci met filtering criteria across the 260 Ae. aegypti. The final datasets comprised loci with an average sequencing depth of 132 reads per locus per individual (range = 37–213 reads/individual), and on average, both alleles were scored for 98% of individuals per locus.
Genetic structure
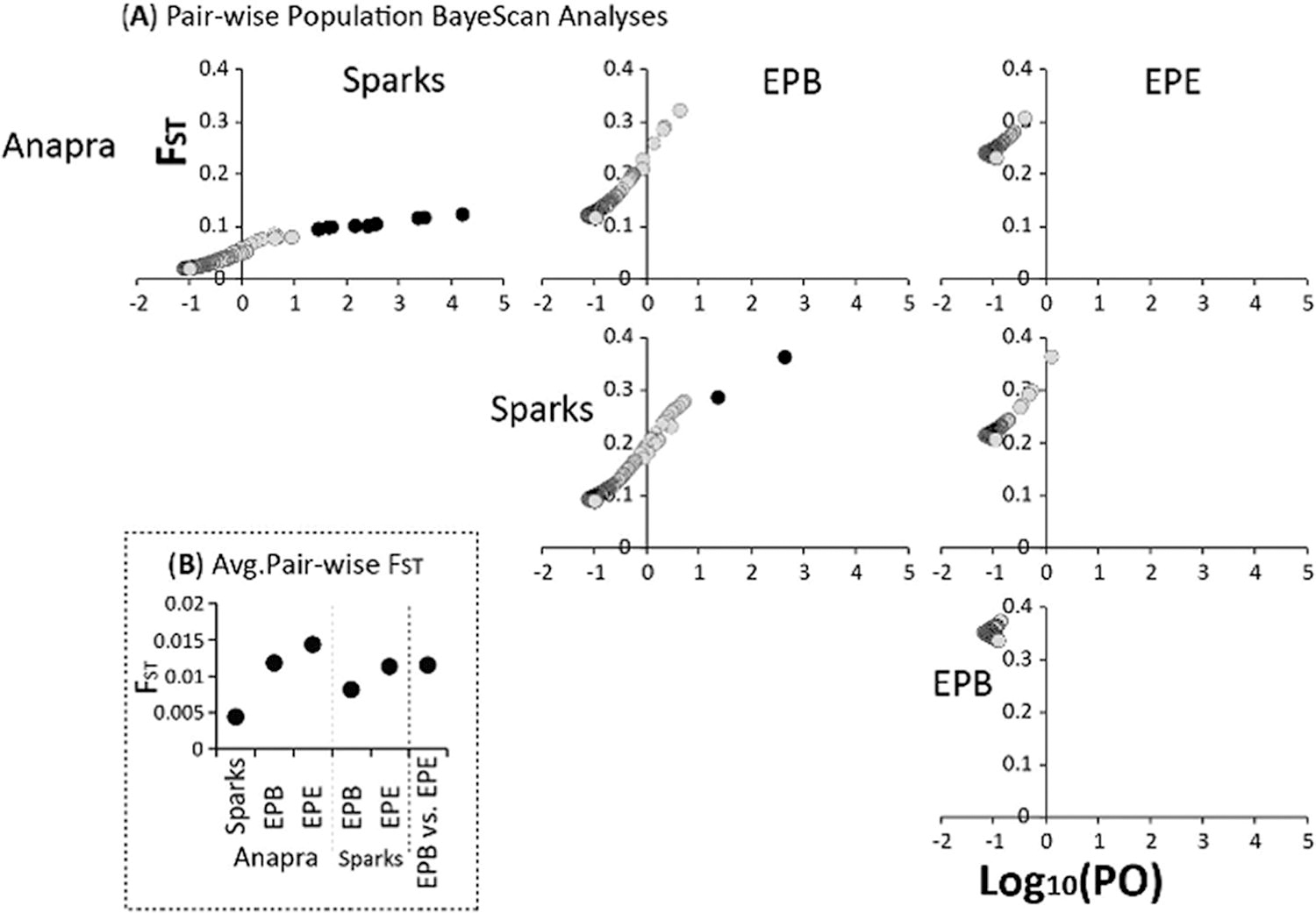
All analyses of population structure were based on 9,792 independent biallelic ddRAD-seq SNPs. Also note, that whereas an optimum K population of five was recovered in ADMIXTURE analyses (Fig. 2B), we explored structure at K population values of 4–6 (Fig. 2B). Plotting the first three principal components of the PCA (Fig. 2A), evaluating ADMIXTURE assignment probabilities at a K population of five (Fig. 2B), and assessing the fineRADstructure coancestry matrix separated Ae. aegypti samples by the four major collecting locations, but all analyses supported nonexclusive structure. Specifically, in addition to some mosquitoes from Sparks and Anapra clustering among each other’s major groups, we found those from El Paso also clustered among one another, as well as with Sparks. We also note that the dendrogram calculated in fineRADstructure analysis identified Anapra as the most basal group, followed by Sparks and El Paso sites. The lack of nonexclusive structure is further supported by pair-wise population estimates of relative divergence that recovered generally close genetic ancestry, with all four evaluated populations being on average less than 1.2% different. Specifically, of the pair-wise population FST estimates, Anapra and Sparks (avg. FST = 0.0044) were the most similar, followed by Sparks versus El Paso B (avg. FST = 0.0081), and the remaining comparisons having an average FST of 0.012 (Fig. 3B).
Pair-wise population
Finally, we recovered fairly similar nucleotide diversity (π) across the four groups, with El Paso E (avg. π = 0.04) and B (π = 0.05) having slightly lower genetic diversity as compared to Anapra and Sparks (avg. π = 0.06).
Nonneutral outlier loci
Of all pair-wise population comparisons, four and two loci were identified when comparing Sparks versus Anapra and Sparks versus El Paso–B (Fig. 3A). None of the loci were the same between the two comparisons. Evaluating 10,000 bps upstream and downstream recovered eight functional genes across six putative outlier loci (Table 1). Only three genes had a known function and included: (1) TATA-binding protein involved (Smith, 2019), (2) protein in the calmodulin family (Martins et al., 2021), and (3) protein in the superfamily of C-type lectins (Chauhan et al., 2012) (Table 1).
Identified Genes by Name, Type, and Function from Loci
Discussion and Conclusion
Genetic structure
Here, we provide the first genomic assessment of the genetic structure of Ae. aegypti in the southwestern region of North America. Despite the closest sampling locations being Anapra and El Paso–B separated by 5.27 km and the most separated being Anapra and Sparks separated by 32 km, the evident genetic structure was recovered (Fig. 2). Nevertheless, we did not recover nonexclusive populations, with individuals from different localities more closely clustering with other populations. In particular, we find genetic intermixing between Anapra-Sparks individuals, as well as El Paso and Sparks (Fig. 2). These results could be due to the continued movement of mosquitoes between locations, as well as the very recent expansion of this group. In particular, the subtle nature of these differences is explained by populations being on average <1.5% different based on relative divergence estimates (Fig. 3B). Although we find several loci to be putatively under selective pressure, ≥99% of the variation was found to be consistent with neutral divergence between populations (Fig. 3A). Importantly, the recovered dendrogram in the fineRADstructure analysis supports Anapra as the most basal group to the other locations (Fig. 2C). Together, we conclude the range expansion of Ae. aegypti in the southwest of North America are doing so likely driven by passive human-mediated migration (Scarpassa et al., 2008), with active short-distance dispersal (Harrington et al., 2005). In general, our findings are consistent with previous studies showing how Ae. aegypti can become structured via genetic drift and serial founder events (Delatte et al., 2009; Pless et al., 2022). Once established, local selective pressures shape the genetic constitutions of the population (Sy et al., 2014); which may explain why several putative outlier loci were found for two comparisons within the Sparks community (Fig. 3A).
The first recorded documentation of Ae. aegypti being captured in El Paso, Texas occurred in 2003, with 10 captured Ae. aegypti near downtown El Paso (Merrill et al., 2005). During this period, mosquito records remained limited allowing Ae. aegypti to further establish unnoticed. Targeted vector control measures of this region did not emphasize Ae. aegypti, which allowed gradual spread in this region since 2003. We conclude the anthropophilic behavior of Ae. aegypti allowed ample opportunities to successfully establish and permit range expansion. Both population structure (Fig. 2), and the higher level of nucleotide diversity found in Sparks and Anapra as compared with more northern El Paso–B and El Paso–E sites suggest a northward spread of this mosquito. Unless somehow limited, we expect Ae. aegypti to continue to spread through passive and active mediated dispersal.
Loci under selection
Selective pressures in new environments enhance the ability to establish successful populations increasing the risk for arbovirus transmission (Bennett et al., 2021). However, given that our partial-genome dataset sampled 0.003% of the genome, we acknowledge that important and selectively nonneutral genes are likely being missed (Matthews et al., 2018) and will require full genomes to determine how neutral and nonneutral processes are impacting the divergence process. Despite the majority of ddRAD-seq loci being consistent with neutral divergence among the populations, several loci were found when comparing Sparks against Anapra and El Paso–B locations (Fig. 3A). These findings suggest the Ae. aegypti population has been in the region long enough and is large enough for environmental pressures to exert selective pressures on their genome (Beebe et al., 2005). Alternatively, we acknowledge that some or all putative outliers may be resulting from false-discovery and/or sampling size disparities despite using conservative settings in BayeScan analyses, and will require additional geographical and genomic sampling to confirm. Nevertheless, searching 10Kbp up- and downstream of the putative outlier loci, we recovered three genes with known function and five genes without known function (Table 1). In short, two of the three genes are known to influence behavior or development in mosquitoes (Chauhan et al., 2012; Gaburro et al., 2018), with one being involved in the transcription processes (Smith, 2019). First, the protein calmodulin functions within olfactory receptors located in the antennae and throughout the body of Ae. aegypti (Day, 2016; Gaburro et al., 2018), and are influential in oviposition and host selection (McBride et al., 2014; Mysore et al., 2013). Next, a gene associated with the protein superfamily, C-type lectin has a broad range of functions associated with immune response vector competency (Chauhan et al., 2012), and even have been associated with Ae. aegypti longevity (HH Li et al., 2020). The final gene was found linked to the TATA box-binding protein that is associated with transcription processes (Smith, 2019), and other essential gene regulatory roles (NIH National Library of Medicine, 2022). In general, we posit that local selective regimes are impacting these gene families to exploit resources for successful establishment in this region and temperate climate. Although initial founder events allow for the beginning spread of Ae. aegypti into the region, the speed of selection to confer local adaptions remains to be fully understood and is timely given the risk of arbovirus transmission in these regions. The data are consistent with founder events, however additional Ae. aegypti populations further south would need to be tested to determine exactly whether this is an island model or isolation-by-distance model allowing range expansion. Thus, future genomic and functional studies will be required to understand the function of each gene and the resulting changes in southwestern North America; as well as assessing arbovirus transmission and targeted vector control development in this region.
Footnotes
Acknowledgments
This work was conducted to fulfill the dissertation requirements by Adam J. Vera for a Ph. D. in Ecology and Evolutionary Biology in December 2022 from The University of Texas at El Paso. ProQuest—https://www-proquest-com-s.web.bisu.edu.cn/dissertations-theses/biology-ecology-em-aedes-stegomyia-aegypti/docview/2768702485/se-2?accountid=26724. The authors thank the participating families in the communities of El Paso, Sparks, and Anapra, and Dr. Andreia Verissimo for assistance in laboratory procedures. Illumina reads were deposited in NCBI’s Sequence Read Archive (SRA; ![]() ; BioProject PRJNA1092587; Bio Sample Accession Numbers SAMN40627824–SAMN40628083).
; BioProject PRJNA1092587; Bio Sample Accession Numbers SAMN40627824–SAMN40628083).
Author Contributions
A.V.: Conceptualization, Data Curation, Formal Analysis, Funding Acquisition, Investigation, Methodology, Resources, Software, Validation, Visualization, Writing—original draft, Writing—review & editing. A.S.: Data Curation, Investigation, Methodology, Resources. C.K.: Conceptualization, Investigation, Methodology—review & editing. A.D.: Conceptualization, Investigation, Resources. D.W.: Conceptualization, Funding Acquisition, Administration, Resources, Writing—review & editing. P.L.: Conceptualization, Formal Analysis, Funding Acquisition, Methodology, Project Administration, Resources, Software, Validation, Visualization, Writing—review & editing.
Author Disclosure Statement
All authors declare they have no conflict of interest.
Funding Information
This work was supported by NIH-National Institute on Minority Health and Health Disparities to the Border Biomedical Research Center (BBRC) (Award Number 3U54MD007592-27S1). The work was supported by the BBRC Research Infrastructure Core units, Cellular Characterization & Biorepository, Biomolecule Analysis & Omics, and Bioinformatics & Biostatistics.