
Abstract
Paramyxoviridae is one of the most well known and largest virus families, including some animal and human pathogens, such as the Hendra, Nipah, and Rinderpest viruses, with a high potential for the emergence of human diseases. Based on recent phylogenetic analyses, two new genera (Narmovirus and Jeilongvirus) have been described. The newly recognized genus Jeilongvirus has rapidly increased in number and has grown to 15 species from 7 a few years ago. However, little is known about the diversity, host range, or evolution of Jeilongvirus. As a well-known host reservoir for many pathogens, rodents have always been the focus for characterizing their pathogenic potential. In this study, we isolated a Tailam virus strain (RN-JH-YN-2022-1) belonging to the genus Jeilongvirus from Rattus norvegicus in Yunnan Province, China. The virus presented a near-complete genome (19,046 nucleotides). Similar to other members of the genus Jeilongvirus, the genome of RN-JH-YN-2022-1 contains eight basic genes (3′-N-P/V/C-M-F-SH-TM-G-L-5′) with 88.88% sequence identity to Tailam virus (TL8K). Additionally, we discuss the pattern of genus Jeilongvirus diversity and the possible route of spread of the Tailam virus, which could provide new clues into the host range, virus diversity, and geographical distribution of the genus Jeilongvirus.
Introduction
Paramyxoviruses (PMVs) are enveloped, negative-strand, nonsegmented RNA viruses belonging to the family Paramyxoviridae, which are divided into 4 subfamilies, 17 genera, and 78 species (Rima et al., 2019). Bats and rodents are the main reservoir hosts of PMVs, and with the increase in the number and host range of samples and improvements in sequencing technology, PMVs have been identified in birds, reptiles, and fish. PMVs that cause human and animal diseases mainly belong to three genera of the subfamily Orthoparamyxovirinae (Henipavirus, Morbillivirus, and Respirovirus), and well-known viruses pathogenic to humans, including Nipah and Hendra viruses, are mainly associated with bat host reservoirs (Chua et al., 2000; Murray et al., 1995).
Due to the rodent species diversity and broad range of distribution, rodent-associated PMVs have also attracted increasing attention as several rodent-borne PMVs have been identified, such as the Nariva virus isolated from forest rodents (Zygodontomys brevicauda) in eastern Trinidad, Mossman virus isolated from wild rats in Australia, and bank vole virus isolated from bank voles (Myodes glareolus) in Russia (Alkhovsky et al., 2018; Lambeth et al., 2009; Miller et al., 2003). As the largest subfamily of PMVs, which are considered most related to infectious diseases, Orthoparamyxovirinae consists of eight genera (Rima et al., 2019).
Based on recent phylogenetic analyses, two new genera (Narmovirus and Jeilongvirus) have been described, a large proportion of which are rodent-associated viruses. Including the J virus, first isolated from mice (Jun et al., 1977), several unclassified rodent- and bat-borne PMVs and members of the genus Shaanvirus were classified into the newly recognized genus Jeilongvirus, which has rapidly increased in number and has grown to 15 species from 7 a few years ago (Rima et al., 2019). However, little is known about the diversity, host range, or evolution of Jeilongvirus.
Yunnan is considered one of the regions richest with the highest animal species worldwide and is also an important channel for many viruses to cross the border from Southeast Asia to China and even East Asia. The aim of this study was to conduct a survey of PMVs among brown rats in Yunnan Province to clarify the natural state and genetic characteristics of the virus in this region and to provide scientific data to support public health prevention and control.
Materials and Methods
A total of 20 rats (10 from Rattus norvegicus and 10 from Rattus tanezumi) were captured in the suburban areas of Mohan Port and Jinghong City, Xishuangbanna Dai Autonomous Prefecture, Yunnan Province, China. All samples were identified based on morphological features, transported with dry ice, and stored in RNA-later at −80°C.
Total RNA was extracted from the lung tissues of the rats using a Tianamp virus RNA kit (Tissue, swab) according to the manufacturer's instructions. PMVs were screened in samples with the gene-specific primers reported previously (Waruhiu et al., 2017). PCR amplification was performed using the One-Step RT-PCR kit (Qiagen, Hilden, Germany). The genome sequence of the positive sample was amplified using primers designed based on the genomes of the Tailam virus and other phylogenic-related PMVs (Supplementary Table S1). Sanger sequencing was performed by Tsingke Biological Co., Ltd. Sequences were assembled and manually edited to produce the final sequences of the viral genomes.
Genome organization was predicted using ORFfinder (https://www.ncbi.nlm.nih.gov/orffinder/) and was confirmed by multiple alignments of the annotated Tailam virus and other related PMV genomes. The amino acid p-distance was calculated to analyze the genetic distance using the amino acid sequences of the N, P, M, F, G (H or HN), L, SH, and TM genes from 18 PMVs belonging to 7 genera (Table 1).
Amino Acid Sequence Similarity (%) Between the Tailam Virus (Jeilong-RN-JH-YN-2022-1) Detected in This Study and Other Paramyxoviruses
The sequences obtained in our study were aligned with reference sequences from GenBank using the ClustalW algorithm with the default parameters. A maximum-likelihood tree was constructed using the MEGA-X program with an appropriate model and 1000 bootstrap replications.
Results and Discussion
Two PMVs were identified in a total of 10 rats belonging to R. norvegicus (2/10), named Jeilong-RN-JH-YN-2022-1 and Jeilong-RN-JH-YN-2022-2. PMVs were not detected in R. tanezumi (0/10). Phylogenetic analysis of the RdRp gene showed that these two PMVs belong to the Tailam virus and cluster with the TL8K strain (Fig. 1).

Phylogenetic analysis based on the amino acid sequences of the L protein of PMVs within subfamily Orthoparamyxovirinae and with Metaavulavirus and Orthorubulavirus as outgroup. The evolution relationships were inferred using the maximum-likelihood algorithm with 1000 bootstraps. Only bootstrap values above 70% are shown. The virus discovered in this study is labeled with red font. PMV, paramyxovirus.
To further elucidate its genetic characteristics, the near-complete genome of Jeilong-RN-JH-YN-2022-1 (19046 bases) was obtained for gene annotation and phylogenetic analysis (accession no. OR573984). The genome of Jeilong-RN-JH-YN-2022-1 contains eight basic genes (3′-N-P/V/C-M-F-SH-TM-G-L-5′) with a G + C content 39.74%. Its genetic structure is mostly similar to the TL8K genome identified from Sikkim rats (Rattus andamanensis) in Hong Kong which is consistent with the “rule of six” as in other PMV genomes.
The phylogenetic tree constructed using the (near) complete genomes of known representative PMVs showed that the 17 reference viruses and RN-JH-YN-2022-1 formed a relatively distinct clade (Fig. 2) belonging to the genus Jeilongvirus harbored by rodents, bats, hedgehogs, and cats.
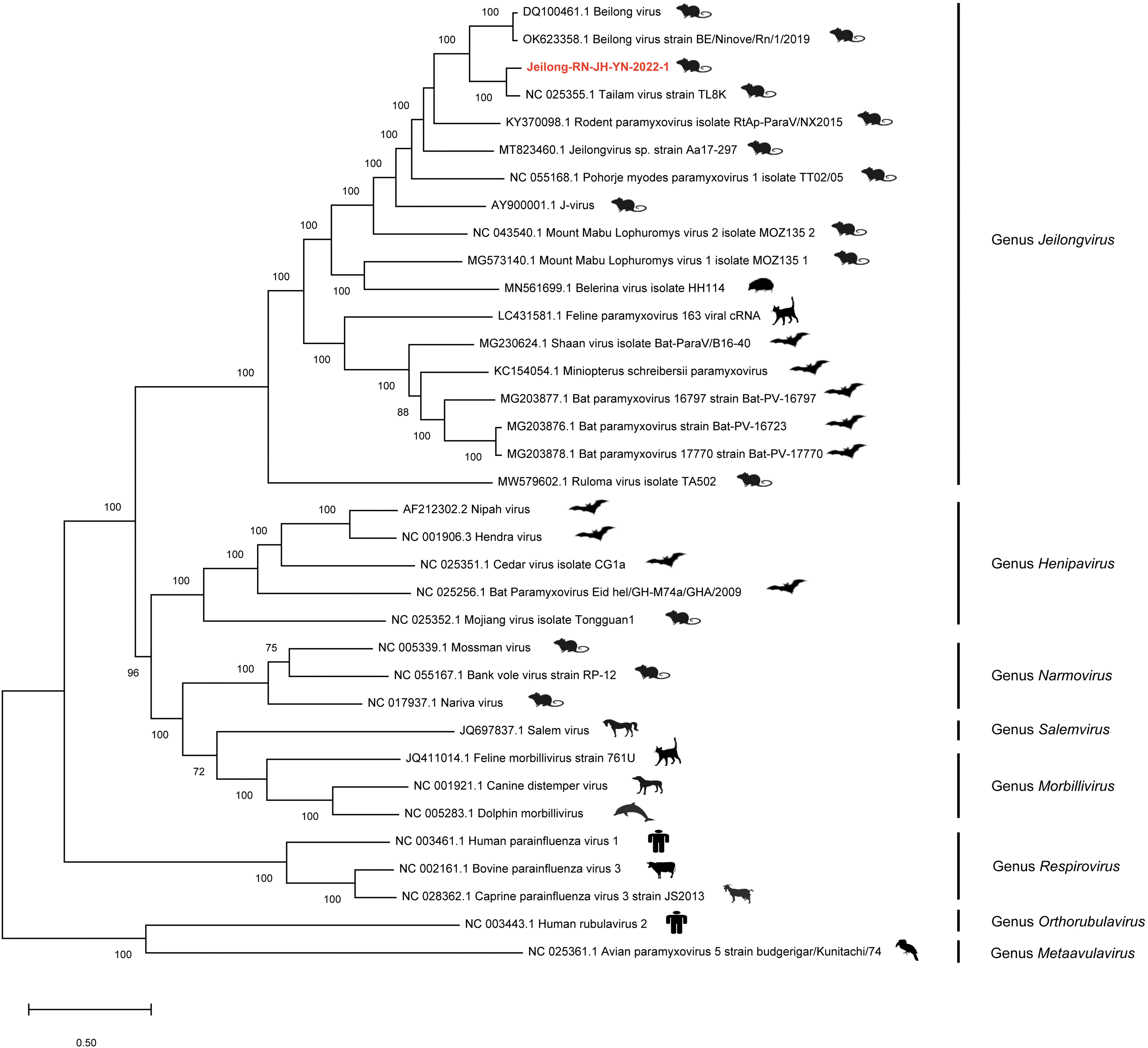
Phylogenetic analysis based on (near) complete-genome sequence of PMVs within subfamily Orthoparamyxovirinae and with Metaavulavirus and Orthorubulavirus as outgroup. The evolution relationships were inferred using the maximum-likelihood algorithm with 1000 bootstraps. Only bootstrap values above 70% are shown. The virus discovered in this study is labeled with red font.
RN-JH-YN-2022-1 was closely related to TL8K detected in Sikkim rats in 2011 and further clustered with the Beilong virus strain BENinoveRn12019 and Beilong virus. More than half of the Jeilong viruses hosted by rodents are distributed worldwide, making them suitable as a model to study the diversity of PMVs.
Pairwise comparisons of the amino acid sequences revealed that YN-2022-1 shared more similarities with members of the genus Jeilongvirus than with other genera (Table 1). Moreover, Jeilong-RN-JH-YN-2022-1 shared the highest similarity with M proteins (ranging from 62.5% to 99.7%) and low similarity with P, SH, and TM proteins within the genus Jeilongvirus. Jeilong-RN-JH-YN-2022-1 showed the highest amino acid identity with TL8K, with N, P/V/C, M, F, SH, TM, G, and L proteins at 98.9%, 93.5%, 99.7%, 97.8%, 96.1%, 94.1%, 87.3%, and 98.3%, respectively, followed by Beilong virus. All amino acid sequence identities of proteins with TL8K exceeded 93%, except for G protein, which was only 87.3%, which is consistent with the fact that G protein shows a higher level of genetic diversity across PMVs (Vanmechelen et al., 2018; Zeltina et al., 2016).
As a new genus described in 2019, the number of Jeilong viruses has increased rapidly, with 15 species constituting the genus to date, as described by the International Committee on Taxonomy of Viruses (ICTV, https://talk.ictvonline.org). Several Jeilong viruses have been isolated since the J virus was first isolated from mice (Jun et al., 1977), including the Beilong virus isolated from a human mesangial cell line (Li et al., 2006) and Shaan virus B16-40 isolated from kidney epithelial cells of an African green monkey (Noh et al., 2018). Recently, a novel PMV, Paju Apodemus paramyxovirus 1 (PAPV-1), was discovered in Apodemus agrarius in the Republic of Korea and was successfully isolated from Vero E6 cells. Furthermore, PAPV-1 was observed to infect human epithelial and endothelial cells and induced the expression of innate antiviral genes (Lee et al., 2021). Continuous surveillance and characterization of potential zoonotic rodent-borne PMVs should be performed because of the increasing risk of cross-species transmission and zoonotic potential.
In this study, we detected a Tailam jeilongvirus strain from R. norvegicus in China for the first time since its discovery in R. andamanensis in 2011. Additionally, a partial sequence of the RdRp gene (KP963900.1) was identified in R. norvegicus in Thailand in 2015 that was predicted to be the Tailam virus; however, we could not conduct the phylogenic analysis because of the lack of complete genome information. Based on the geographic origin, migration, and strong adaptability of R. norvegicus (Zeng et al., 2018), the distribution of Tailam virus in R. norvegicus may be related to the migration of the host from Southern East Asia to Northern East Asia and was introduced to R. andamanensis through the host switch. Additionally, although Tailam viruses are phylogenetically closely related to the Beilong virus, the range of host species and virus prevalence are much lower. The difference in host specificity between Tailam and Beilong viruses is possibly associated with the low amino acid sequence identity of the G protein (65%), which plays a significant role in cell entry through the binding of the virus to host target cells and is likely to be a major determinant of PMVs host specificity (Zeltina et al., 2016).
In summary, rodent-borne PMVs have drawn more public health attention because of the potential of leading to disease emergence since the first rodent-borne Henipavirus Mòjiang virus was identified in pneumonia cases in China in 2012, especially for the new genus Jeilongvirus, which nearly accounts for half of all known orthoparamyxoviruses. Jeilongvirus genomes contain more variable genome lengths (from 14.8k to 20.5k) compared to other genera, and they harbor one or two additional genes (TM and SH) encoding membrane-associated proteins that are related to cell-to-cell fusion in the J virus (Li et al., 2015). However, the precise functions of the TM and SH genes across the whole genus remain unclear.
In addition, it is worth noting that frequent host exchanges between introduced and endemic small rodents play a significant role in increasing the infection levels of Jeilongvirus apodeme in Madagascar (Wilkinson et al., 2014), and we do not know if a similar situation occurs with other viruses. Additionally, rats (Lophuromys machangui) in Mozambique were coinfected with two species of Jeilong virus (Mount Mabu Lophuromys virus 1, 2) (Vanmechelen et al., 2018). However, studies on the diversity and host range of most Jeilong viruses are mainly based on the identification of a short fragment of the RdRp gene, and more genomic information on the virus is needed to understand how key forces such as virus-host codivergence and cross-species transmission drive virus diversity and evolution.
Institutional Review Board Statement
The study was approved by the Institutional Ethical Committee of Kunming University of Science and Technology (protocol no. 16,048).
Data Availability Statement
The data presented in this study are openly available in the NCBI database by accession number OR573984.
Footnotes
Authors' Contributions
Conceptualization, Y.F., Y.H., B.W., and Xueshan Xia; methodology, Y.F. and B.W.; software: Y.M., Q.L., X.L., and J.J.; formal analysis: Y.F., Y.H., and Xiang Xu; writing—original draft preparation, Y.F.; writing—review and editing, Y.F., B.W., and Xueshan Xia; funding acquisition, Xueshan Xia and B.W. All authors have read and agreed to the published version of the manuscript.
Author Disclosure Statement
No conflicting financial interests exist.
Funding Information
This study was supported by Yunnan Key R&D Program (grant no. 202103AQ100001), Yunnan Major Scientific and Technological Projects (grant no. 202202AG050013), Yunnan Province Basic Research Program Projects (grant no. 202101AS070028).
Supplementary Material
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.