
Abstract
Background:
Chikungunya is a zoonotic disease caused by the Chikungunya virus (CHIKV), primarily transmitted to humans through infected Aedes mosquitoes. The infection is characterized by symptoms such as high fever, musculoskeletal pain, polyarthritis, and a rash, which can lead to severe complications such as encephalitis, meningitis, and even fatalities. While many disease manifestations resemble those of other viral infections, chronic arthritis caused by CHIKV is unique, and its molecular mechanisms remain ill-defined.
Materials and Methods:
Proteomics data from both cellular and patient levels of CHIKV infection were curated from PubMed and screened using inclusion and exclusion criteria. Patient serum proteomics data obtained from PRIDE underwent reanalysis using Proteome Discoverer 2.2. Enrichment and protein-protein interaction network analysis were conducted on differentially expressed proteins from both serum and cellular datasets. Metabolite data from CHIKV-infected patients were further retrieved, and their protein binding partners were identified using BindingDB. The protein-metabolite interaction pathway was further developed using MetaboAnalyst.
Results:
The proteomics data analysis revealed differential expression of proteins involved in critical host mechanisms, such as cholesterol metabolism and mRNA splicing, during CHIKV infection. Consistent upregulation of two actin cytoskeleton proteins, TAGLN2 and PFN1, was noted in both serum and cellular datasets, and their upregulations are associated with arthritis. Furthermore, alterations in purine metabolism were observed in the integrative proteome-metabolome analysis, correlating with cytoskeletal remodelling.
Conclusion:
Collectively, this integrative view sheds light on the involvement of actin cytoskeleton remodeling proteins and purine metabolic pathways in the development of arthritis during CHIKV infection.
Introduction
Chikungunya infection, which is caused by the Chikungunya virus (CHIKV) and primarily transmitted by Aedes mosquitoes, including Aedes aegypti and Ae. albopictus, was initially identified in Tanzania during the early 1950s (Mason and Haddow, 1957; Singh and Unni, 2011). CHIKV belongs to the Alphavirus genus within the Togaviridae family and possesses a single-stranded, positive-sense RNA genome. This is ∼11.8 kb long and contains two open reading frames (ORF) that encode viral proteins. The ORF located at the 5′ end encodes four nonstructural proteins (nsP1, nsP2, nsP3, and nsP4), whereas the ORF at the 3′ end encodes five structural proteins (capsid C, E1, E2, E3, 6K, and the transframe protein TF) (Patil et al., 2018). After entering host cells, the RNA is translated into a polyprotein, which is subsequently cleaved into nonstructural proteins (nsPs) responsible for viral replication and structural proteins, such as capsid (C) and envelope glycoproteins (E1 and E2), hence facilitating viral assembly (Ahola et al., 2015; Rausalu et al., 2016; Zhang et al., 2019). Chikungunya fever caused by CHIKV is characterized by a sudden onset of fever, polyarthralgia (pain in multiple joints), and a maculopapular rash. The incubation period typically spans 2–4 days, and asymptomatic infections are observed in 5–15% of cases. Although CHIKV infection is generally nonfatal, it is noteworthy that more than 40% of patients develop arthritic symptoms that persist for over 3 months after the acute illness, termed “chronic chikungunya arthritis” (Amaral et al., 2020; Gasque et al., 2015).
In India, CHIKV continues to evolve, driven by the country’s vast population, a significant number of immunologically naive individuals, and the presence of abundant Aedes mosquitoes (Translational Research Consortia for Chikungunya Virus in India, 2021). Notably, unique genetic changes, particularly in the E2 protein coding region, have been observed in Kerala (Niyas et al., 2010). Besides, whole genome sequencing studies conducted in eastern India have uncovered microevolutionary trends within CHIKV, resulting in strains with modified virulence and increased transmission potential (Dutta et al., 2018; Nath et al., 2023). In addition, a study conducted in Delhi, India, revealed co-infections of CHIKV and DENV (Dengue virus) in 10% of the study sample and strains in that locality clustered within the East–Central–South East African genotype (Afreen et al., 2014). Molecular characterization of chikungunya virus in the north Indian population revealed significant mutations in amino acids of both nonstructural and structural proteins, affecting hydropathicity, isoelectric point, and instability index (Khan et al., 2021). Recent research conducted in Kerala made a significant breakthrough by identifying a novel mutation in the E2 gene of CHIKV strains circulating between 2014 and 2019, shedding light on the ongoing molecular evolution of CHIKV (Anukumar et al., 2021).
CHIKV-infected patients exhibit a range of rheumatic symptoms, including symmetrical polyarthritis, resembling rheumatoid arthritis, nonspecific arthralgias akin to postviral arthritis, and asymmetrical oligo or mono arthritis similar to seronegative spondyloarthritis. Other potential outcomes encompass postviral polyarthralgia, chronic joint pain, fibromyalgia, adhesive capsulitis, and plantar fasciitis (Bouquillard and Combe, 2009; Javelle et al., 2015). Extensive research has explored mechanisms connecting viral infections to the development of rheumatic diseases. Molecular mimicry is one such mechanism, where similarities between host antigens and viral proteins disrupt immune tolerance, resulting in arthritis characterized by elevated cytokine signaling and increased proinflammatory factors, such as interleukin-1 (IL-1), IL-6, and Tumor necrosis factor alpha (TNF-α) (Kany et al., 2019). Alternatively, viral infections can induce dysfunction in T and B cells, potentially triggering autoimmune responses in genetically susceptible individuals (Smatti et al., 2019). Comparing the pathogenesis of chronic chikungunya arthritis with rheumatoid arthritis reveals shared mechanisms, including elevated levels of proinflammatory cytokines, such as IL-6, Granulocyte macrophage colony-stimulating factor (GM-CSF), Interferon alpha (IFN-α), and IL-17 (Amaral et al., 2018).
In addition to the typical inflammatory response, we wanted to gain a deeper understanding of the mechanisms underlying joint pain and arthritis resulting from chikungunya infection. Therefore, a comparative analysis of the proteome–metabolome datasets was conducted. This pilot study considered various samples, including serum and cell lines, affected by CHIKV Indian variant. Proteins with shared differential expression patterns in patient serum samples and virus-infected cell lines were identified, and their involvement with multiple pathways were analyzed. Subsequently, a proteome–metabolome network was constructed to unveil the potential molecular networks triggered by CHIKV infection.
Materials and Methods
Proteomics data analysis and identification of differentially regulated proteins
To analyze protein-level variations during CHIKV infection, we conducted a comprehensive literature survey on PubMed using the MeSH terms “Chikungunya” OR “CHIKV” AND “proteomics” NOT “review.” The curated articles underwent rigorous screening based on predefined inclusion and exclusion criteria to standardize the data. Our focus was to primarily capture the articles containing proteomics datasets at the patient serum and human cellular levels following CHIKV infection in the Indian population, along with their corresponding control samples.
The data pertaining to serum proteomics of patients affected by chikungunya were downloaded from ProteomeXchange database through PRIDE partner repository (PXID: PXD000234) (Puttamallesh et al., 2013). In this study, control serum samples from four healthy individuals and chikungunya samples from five infected cases were collected. Equal amount of proteins from each individual were pooled into two groups: the control and infected. Control samples were labeled with iTRAQ-114 reagent, whereas infected samples were labeled with iTRAQ-115. These samples were pooled and fractionated into 22 fractions and analyzed by mass spectrometry. Therefore, the data had 22 files, which were reanalyzed using Proteome Discoverer 2.2 (Thermo Scientific, Bremen, Germany) with SEQUEST HT and MASCOT algorithms using reporter ion quantification (Eng et al., 1994; Koenig et al., 2008). The data were searched against Reference human proteome from NCBI-RefSeq database (Version 109, 111,452 entries). Up to one missed cleavage was allowed for trypsin. Precursor and fragment mass tolerance was set to 10 ppm and 0.6 Da, respectively. Oxidation of methionine, acetylation of N-terminus, and iTRAQ modification at the N-terminus were searched as dynamic modifications. Methythiol modification of cysteine and iTRAQ modification on lysine were searched as static modifications. Peptide–spectrum matches (PSMs), peptides, and proteins were identified at a false discovery rate (FDR) of 1%. After the identification of proteins, the raw abundances were normalized by smooth quantile normalization method. For both processed cellular data and reanalyzed serum data, we identified differentially regulated proteins (DRPs) with an absolute fold change cutoff of ∓1.3 with a p-value <0.05.
Enrichment analysis of differentially regulated proteins
Gene ontology (GO) enrichment analysis was performed separately for both the serum and the cellular differentially expressed proteins (DEPs) using the online ShinyGO version 0.77. This provided information on GO categories, encompassing cellular components (CC), molecular function (MF), and biological process (BP), as well as top enriched pathways retrieved from the KEGG database (Ge et al., 2020; Kanehisa and Goto, 2000). To enhance the significance of the enrichment analysis, a FDR cutoff of 0.05 was maintained, against the species “Human.”
Differentially regulated proteins network analysis
The protein–protein interaction (PPI) network of differentially regulated proteins was constructed using the Search Tool for the Retrieval of Interacting Genes database. For enhanced significance, the active interaction source, text mining, was turned off (Szklarczyk et al., 2021). Subsequently, this network was imported into Cytoscape (version 3.10.0) for further analysis (Shannon et al., 2003). To identify functional clusters within the network, the Molecular Complex Detection (MCODE) plugin in Cytoscape was deployed. MCODE identifies these clusters by evaluating nodes based on their local neighborhood density and expands its analysis outward from densely connected seed proteins to uncover larger, densely interconnected regions, thus revealing functional modules within the network (Bader and Hogue, 2003). In addition, the CytoNCA plugin in Cytoscape was used to identify hub proteins in the biological network (Tang et al., 2015). In CytoNCA, the degree parameter (without weight) was utilized to analyze the network, where a higher degree value indicated a greater number of interactions with other proteins.
Proteome–metabolome integrative network analysis
CHIKV-infected patients’ serum metabolites were collected from the public literature domain, PubMed. Toward this, we used the following MeSH terms, “Chikungunya” OR “CHIKV” AND “metabolomics” NOT “review.” The protein-binding partners of the metabolites were identified using BindingDB, a protein–small molecule interaction database based on experimental evidence (Gilson et al., 2016). To retrieve the interacting target proteins, we used a metabolite similarity score of 0.85 or higher, with hits achieving a score of 1.0 considered as precise matches. The metabolites were converted to their respective SMILES IDs for this analysis. Furthermore, an integrated pathway of protein–metabolite interactions was created using MetaboAnalyst suite version 5.0 (Pang et al., 2021). This analysis utilized the predicted protein targets from BindingDB and the DEPs identified from the serum data after CHIKV infection for generating the integrative network.
Results
Assembly of serum- and cellular-level proteins perturbed by CHIKV infection
To evaluate the extent of proteomic changes during chikungunya infection, we examined the proteomics data available in existing literature at both the patient serum level and also in individual cell lines. The proteomic dataset published by our team is the only study related to patient samples available in the PubMed. The serum samples analyzed in this study were obtained from an Indian population consisting of five infected individuals and four age and sex-matched healthy controls. Our literature survey yielded five articles for mass spectrometry-based proteomics datasets on Indian variant CHIKV infection in human cell lines. With the limitations in the availability of datasets, we narrowed our focus to a more specific context and selected cellular datasets of CHIKV infection using variants of virus found in India that are compatible with the population of the serum-level proteomics data. This stringent scrutiny led to the inclusion of two cellular-level datasets: one from HEK293 cells and another from U-87 MG cells. We excluded the dataset from the U-87 MG cells, specifically, being a malignant glioma cancer cell line. Following the inclusion and exclusion analysis of the assembled data, we proceeded with the serum dataset and the HEK-293 cellular dataset for further analysis (Abraham et al., 2015; Puttamallesh et al., 2013).
As the serum-based dataset was publicly available and to maintain consistency in data analysis parameters with the cell line proteomics datasets, we conducted a reanalysis of the serum proteomics data. In total, we identified 38,549 PSMs from 112,419 MS/MS spectra, corresponding to 4705 peptide groups belonging to 2191 human proteins. Among these proteins, we identified 55 DRPs, with 34 proteins showing upregulation and 21 proteins showing downregulation. In HEK293 cells, among the 106 DRPs, 96 and 10 proteins were reported to be up- and downregulated, respectively, in response to CHIKV infection.
Functional enrichment analysis of CHIKV-induced DRPs in serum and cell lines
To establish the function significance of DRPs identified in the patient serum and cell line datasets, we conducted separate functional enrichment analysis. The top 10 enriched BP, CC, and MF were examined to determine the characteristics of DRPs.
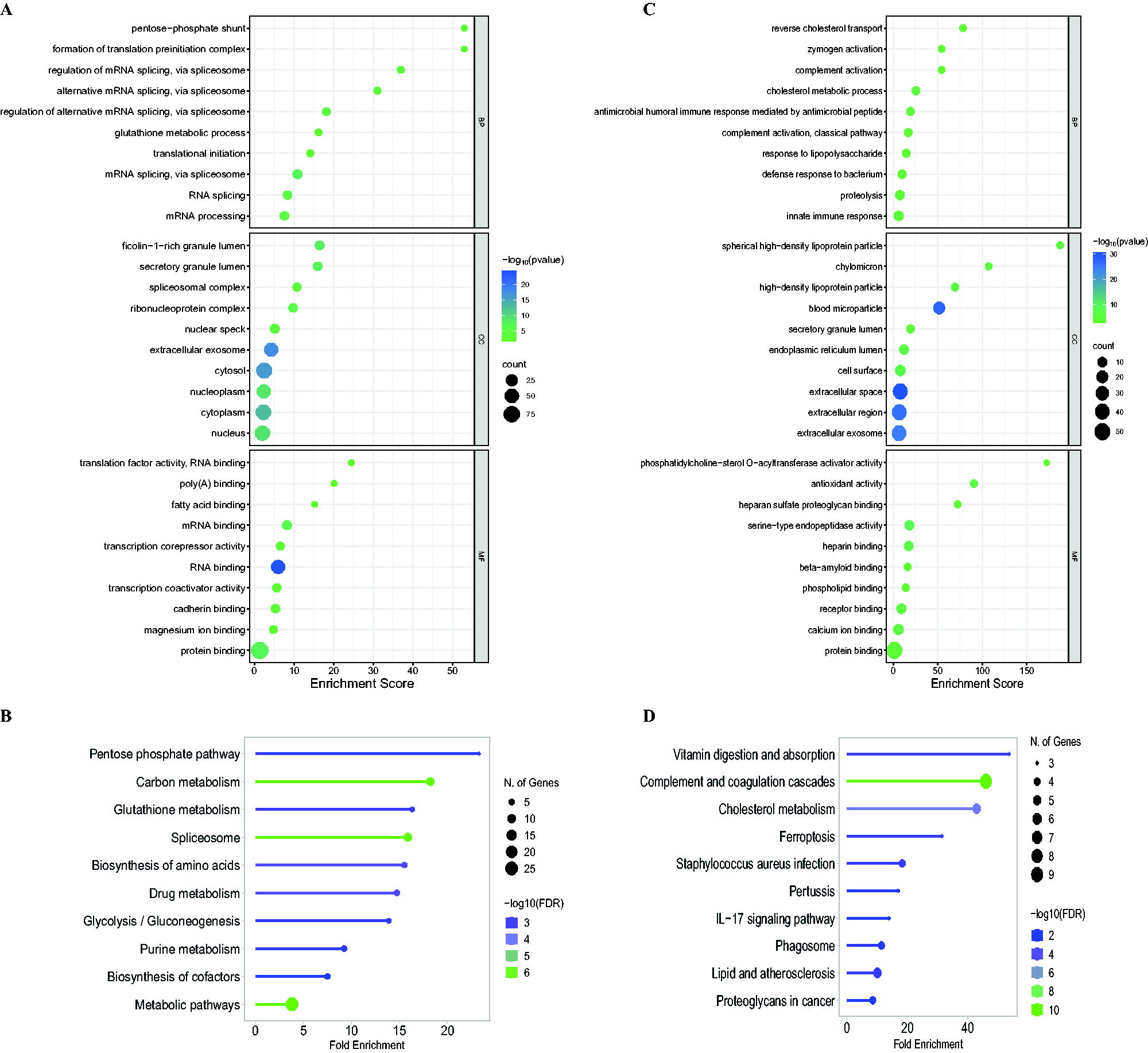
In the cellular dataset, top enriched pathways among the DRPs included the pentose phosphate pathway, carbon metabolism, and the spliceosome pathway. The enrichment of the pentose phosphate pathway may be attributed to the virus’ efforts to reconfigure host cell metabolism to enhance its replication and dissemination, potentially leading to increased NADPH production and metabolic reprogramming. Similarly, the enrichment of the spliceosome pathway suggests the virus’ influence on host gene expression, essential in RNA splicing and modulation. Collectively, these enriched pathways indicate alterations in host metabolism (Fig. 1B).
Proteomic interpretation of differential enrichment during CHIKV infection.
In contrast, the patient dataset showed top enrichment in pathways, such as vitamin digestion and absorption, complement and coagulation cascades, and cholesterol metabolism. Notably, the enrichment of proteins involved in cholesterol metabolism and lipid pathways aligns with literature suggesting that certain viruses can reshape lipid metabolism, boosting cholesterol production to aid viral replication. These enrichments highlight the complex nature of viral infections, potentially leading to complications. The variations in enrichment between the two datasets can be attributed to the differing nature of serum samples compared with cell lines (Fig. 1D).
Consistent with the pattern of enrichment, the top biological processes enriched with cellular data highlight alterations in spliceosome-mediated mRNA splicing. Meanwhile, the top enrichments in molecular functions highlight alterations in binding interactions, such as mRNA, protein, and cadherin. Similarly, the top biological processes enriched with serum data highlight alterations in complement activation and cholesterol metabolism, whereas the top enrichments in molecular functions mirror those seen in cellular data. This includes alterations in binding interactions, such as heparin and phospholipid, among others. In addition, there are alterations in phosphatidylcholine-sterol O-acyltransferase activation, which is essential in cholesterol metabolism. These alterations align with the notion that CHIKV is capable of strategically replicating effectively by evading host defenses (Fig. 1A, C).
Network construction and module analysis
To gain a better understanding of altered protein complexes during CHIKV infection, we constructed PPI networks for the DEPs in both cellular and serum data. String DB was used to generate the PPI networks and subsequently analyzed them in Cytoscape. In the cellular data network, which comprised 86 nodes and 282 edges (Fig. 2A), we identified two significant clusters using the Cytoscape plugin MCODE. The first cluster had a cluster score of 9 and consisted of 9 nodes and 36 edges (Fig. 3A), whereas the second cluster had a cluster score of 5.09 and encompassed 12 nodes and 28 edges. We found that proteins in both clusters were upregulated and functionally significant in mRNA processing and splicing, specifically through the spliceosome pathway. Hub proteins, which are highly connected units within a PPI network that coordinate various cellular processes, were further analyzed using the degree without weight algorithm in the Cytoscape plugin CytoNCA. Our analysis revealed that the top three hub proteins, namely SRSF1, SYNCRIP, and SRSF3, had degrees of 23, 20, and 19, respectively, and were all part of the initial cluster. Notably, all three hub proteins play a role in the regulation of mRNA splicing.

Protein–protein interaction network with DRPs during CHIKV infection
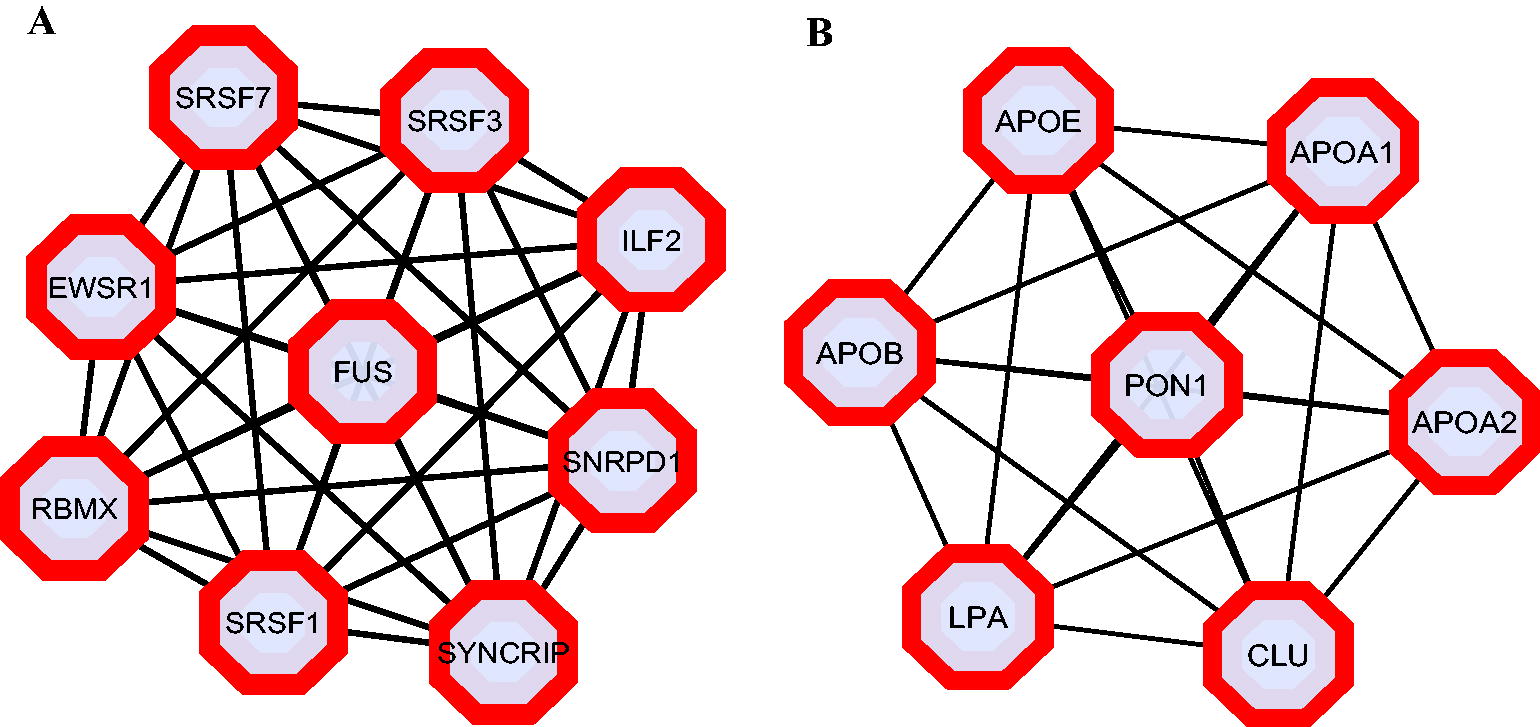
Cluster analysis of PPI network with DRPs during CHIKV infection.
Similarly to the serum data network, which included 38 nodes and 76 edges (Fig. 2B), our analysis identified a single cluster with significant modular characteristics. This cluster, with a cluster score of 7, consisted of 7 nodes and 21 edges (Fig. 3B), and all proteins within it were upregulated. In addition, these proteins demonstrated functional significance in cholesterol metabolism. The top three hub proteins in the network APOB, LPA, and APOA1, each with degrees of 12, 11, and 11, respectively, were part of this cluster.
Comparative proteome analysis
In our quest to bridge proteomic insights between cellular and patient levels data after CHIKV infection, we compared DRPs identified in serum samples with those from cellular datasets, using uniform criteria. The analysis identified two proteins, TAGLN2 (Transgelin 2) and PFN1 (Profilin 1), present in both datasets, exhibiting identical expression patterns. Both TAGLN2 and PFN1 were upregulated in serum samples and cell lines following CHIKV infection. TAGLN2 is a vital protein primarily found in smooth muscle cells, playing a central role in maintaining cellular structure, regulating smooth muscle contraction, and influencing processes, such as cell adhesion, movement, and tissue remodeling, by stabilizing actin structures. PFN1, in contrast, is a multifaceted actin-binding protein that centralizes cellular movement, structure, and signaling by guiding actin filament elongation, thereby affecting essential processes such as cell migration, division, and internalization.
The alterations in actin dynamics observed during CHIKV infection are noteworthy since both of these upregulated proteins are involved in actin dynamics. CHIKV primarily enters cells through clathrin-mediated endocytosis. Given the crucial role of actin in clathrin-mediated endocytosis, which includes aiding in membrane curvature, cargo selection, vesicle scission, vesicle transport, and overall membrane remodeling, the upregulation of actin dynamics could facilitate the viral entry and intracellular transport of CHIKV particles through actin-based motor proteins. However, it is important to note that actin-binding proteins may also be upregulated as part of the host cell's antiviral response. These proteins could potentially play a role in defending the cell against viral infection or repairing cellular damage caused by the virus.
Integrative proteome–metabolome network analysis
To gain a deeper understanding of the pathophysiology of CHIKV infection, we conducted an integrative omics analysis using metabolomics data from 15 CHIKV-infected patients in India (Shrinet et al., 2016). This study identified 10 downregulated and 25 upregulated metabolites during CHIKV infection. To uncover the interactions between the proteome and metabolome during CHIKV infection, we constructed an integrative network using metabolites and their corresponding target protein information through MetaboAnalyst 5.0. This network includes 7 metabolites and 36 proteins (Fig. 4). In the subsequent enrichment analysis, purine metabolism emerged as the top significant pathway, with a p-value of 0.00113. This pathway involves four proteins: catalase (CAT), apolipoprotein E (APOE), PDE4A, and VEGFA, along with one metabolite, theophylline.

Integrative proteome–metabolome interactive network with differentially regulated proteins and metabolites during CHIKV infection. Proteins that interact with multiple metabolites are color-graded differently.
Discussion
Current proteomics strategies have gained prominence because of their ability to offer comprehensive proteome coverage and improved spatiotemporal resolution, made possible by advanced mass spectrometry platforms. The comparative proteomics analysis impact during CHIKV infection reveals the viral strategies used to manipulate various protein components, enabling the virus to evade the host’s defense mechanisms following viral entry and facilitating its replication. From the reanalyzed patient-level data, we observed an enrichment of proteins associated with cholesterol metabolism and lipid pathways in both the gene ontology analysis and pathway enrichment analysis. This consistent pattern of functional significance, centered around cholesterol metabolism, was also identified in the PPI network analysis. In the PPI network, proteins linked to cholesterol metabolism formed the top cluster, all of which showed upregulation. It is worth noting that several enveloped viruses, such as SARS-CoV-2, hepatitis C virus (HCV), human cytomegalovirus, and Epstein–Barr virus, are known to exploit cholesterol metabolism to acquire raw materials for virus particle replication, assembly, and maturation (Dai et al., 2022; Del Campo and Romero-Gomez, 2015; Lange et al., 2019; Syed et al., 2010). Given that CHIKV is also an enveloped virus with viral particles enclosed within icosahedral capsids, the enrichment of proteins related to cholesterol metabolism and lipid pathways strongly suggests that CHIKV may similarly hijack cholesterol metabolism to facilitate its replication. This notion is supported by proteome analysis conducted on HFLS cells, which have shown alterations in proteins associated with host cell immune response, cytoskeletal organization, protein modification, and metabolic processes during CHIKV infection (Sukkaew et al., 2020).
The cellular data revealed a significant enrichment of proteins associated with mRNA processing and splicing, particularly through the spliceosome pathway. This consistent finding was also observed in the protein–protein network analysis. The prominent cluster identified in the network analysis consisted of proteins associated with mRNA splicing, including hub proteins, such as SRSF1, SYNCRIP, and SRSF3, all of which were upregulated during CHIKV infection. Alphaviruses, including CHIKV, induce ER stress during infection, leading to the activation of the unfolded protein response (UPR) through the Inositol-requiring enzyme type 1 (IRE-1) branch. This results in the splicing of XBP1 mRNA, which triggers the expression of genes involved in protein degradation (Ahola and Merits, 2016). However, studies have indicated that CHIKV uses a strategy to evade the UPR by utilizing its nsP2 and nsP4 proteins. These viral proteins interfere with XBP1 mRNA splicing, leading to incomplete splicing and preventing the full activation of UPR-associated genes. This viral evasion mechanism allows CHIKV to manipulate host cell processes to its advantage during infection, as supported by existing literature (Fros et al., 2015; Rathore et al., 2013). Therefore, the enrichment of mRNA splicing-associated proteins during CHIKV infection is likely a consequence of these intricate host–virus interactions.
GO analysis of both serum and cell line samples has revealed an enrichment of complement activation mechanisms. Complement activation plays a crucial role in the development of arthritis, particularly in autoimmune variants such as rheumatoid arthritis and systemic lupus erythematosus (Dijkstra et al., 2019). This complex system served as a first-line defender against infections and cellular damage. In the context of arthritis, complement activation initiates a series of events that result in the recruitment and activation of immune cells, thus contributing to persistent inflammation and tissue damage within the affected joints. The process involves the generation of inflammatory mediators such as C3a and C5a, which attract immune cells, such as neutrophils and macrophages, to the inflamed region. Furthermore, in autoimmune arthritis, the formation of immune complexes because of the erroneous recognition of self-antigens exacerbates complement system activation (Triggianese et al., 2023). Within the synovial fluid of afflicted joints, complement activation sustains inflammation, leading to the release of proinflammatory cytokines and continuous tissue damage. In addition, the creation of the membrane attack complex has the potential to directly harm synovial tissue and cartilage, further complicating the pathology of arthritis (Sturfelt and Truedsson, 2012).
In our comparative proteome analysis of cellular- and patient-level data, we identified two proteins that exhibited a consistent pattern of upregulation in both datasets: TAGLN2 and PFN1. These proteins are essential components of the actin cytoskeleton, contributing to the maintenance of cellular structure and movement (Carlsson et al., 1977; Huang et al., 2021; Shapland et al., 1993;Witke, 2004). Given that CHIKV employs clathrin-mediated endocytosis for host cell entry, the upregulation of TAGLN2 and PFN1 assumes significance, as actin and its cytoskeleton proteins play vital roles in facilitating membrane curvature, vesicle scission, transport, and overall membrane remodeling during clathrin-mediated endocytosis (Loebrich, 2014; Mooren et al., 2012). Furthermore, PFN1 plays a pivotal role in regulating actin dynamics. It promotes polymerization at low concentrations while inhibiting it at high concentrations, thereby exerting control over cell motility, migration, and actin stability through its interaction with actin barbed ends (Courtemanche and Pollard, 2013; Pernier et al., 2016). PFN1 expression is consistently upregulated in various pathological conditions, including psoriasis (both in skin and serum) (Mok et al., 2022), osteoarthritis (associated with bone marrow lesions) (Shabestari et al., 2020), psoriatic arthritis (present in synovial fluid) (Cretu et al., 2014), and atherosclerosis (noted in atherosclerotic plaques and serum) (Caglayan et al., 2010). These upregulations often correlate with disease severity. In our study, we also observed a common upregulation of PFN1 in cellular and serum data following CHIKV infection, suggesting its potential involvement in disease symptoms. For instance, the joint pain seen in conditions such as osteoarthritis, psoriatic arthritis, and chikungunya infection may be linked to the increased expression of PFN1, which is a shared feature among these conditions.
Recent studies have reported that CHIKV relies on actin polymerization for the release of its progeny, involving the activation of MK2 and MK3. In addition, the study emphasized that CMPD1, an MK2 inhibitor, has the potential to suppress viral infection by impeding actin-related processes. By inhibiting MK2/3 phosphorylation, CMPD1 sustains active Cofilin, which cleaves actin polymers into monomers, thereby hindering lamellipodium formation and the release of viral progeny (Mamidi et al., 2021). The upregulation of PFN1 and TAGLN2, as identified in our analysis, holds significance in this context. PFN1 is recognized for enhancing the propulsion speed of lamellipodial protrusion in bacterial pathogens, resembling cell membrane protrusion (Ding et al., 2012). On the contrary, TAGLN2 is known for its colocalization with actin stress fibers and its presence at the lamellipodial leading edge, where active actin polymerization occurs (Na et al., 2015). In addition, TAGLN2 has been demonstrated to induce filopodia-like membrane protrusions at the lamellipodial edge in various cell types (Kim et al., 2018). The molecular characteristics of TAGLN2 suggest its crucial role in inducing small filopodia formation and membrane ruffling, indicating its significance in CHIKV progeny release (Kim et al., 2021). Filopodia act as sensors, aiding the cell in navigation and detecting cues from its surroundings, while lamellipodia provide the necessary forward movement and stability for effective cell migration. Collectively, these studies highlight the importance of the upregulated PFN1 and TAGLN2 identified in our analysis in the context of CHIKV progeny release.
Furthermore, the proteome–metabolome integrative analysis highlighted altered proteins and metabolites associated with purine metabolism during CHIKV infection. Studies have reported that dysregulated purine synthesis plays a crucial role in chronic inflammation and tissue damage seen in various forms of inflammatory arthritis. In the case of CHIKV infection, the virus triggers an inflammatory response in the joints, leading to symptoms resembling inflammatory arthritis. This illustrates how CHIKV infection can result in arthritis. In addition, a study conducted on human fibroblasts reported that purine-induced Ca2+ signaling was associated with actin cytoskeleton remodeling (Goldman et al., 2013). Furthermore, the observed downregulation of CAT and upregulation of APOE indicate an increase in oxidative stress and broader cellular effects, including lipid and cholesterol transport, which are vital components of purine metabolism, further underscoring their significance in this context. Altogether, the study explored the impact of chikungunya infection at the proteome and metabolome level and identified the role of actin-based cytoskeleton remodeling in CHIKV-mediated arthritis. Further experimental validations are required to provide substantial evidence supporting the finding of the present study.
Conclusions
In summary, this study offers valuable insights into the pathophysiology of CHIKV infection through the application of an integrative proteomic–metabolomic data analysis approach. The analysis successfully identified differentially expressed proteins during CHIKV infection, impacting critical host mechanisms, such as cholesterol metabolism and mRNA splicing, through changes in the expression levels of associated proteins. Alterations in purine metabolism, as identified in the proteome–metabolome integrative analysis, correlate with modified cytoskeletal remodeling and the polyarthralgia symptoms, swelling, and joint pain observed in CHIKV infection. In addition, the study revealed the consistent upregulation of two actin cytoskeleton proteins, namely TAGLN2 and PFN1, in both serum and cellular datasets. This persistent upregulation of PFN1, TAGLN2 has been associated with various pathological conditions, including arthritis. Collectively, this research sheds light on the involvement of actin cytoskeleton remodeling proteins in the development of arthritis during CHIKV infection. To further advance our understanding of the roles played by PFN1 and TAGLN2 and purine metabolism in CHIKV-mediated arthritis, additional research should be conducted.
Footnotes
Acknowledgments
The authors thank Karnataka Biotechnology and Information Technology Services, Government of Karnataka, India, for the support to Center for Systems Biology and Molecular Medicine at Yenepoya (Deemed to Be University) under the Biotechnology Skill Enhancement Program in Multiomics Technology (BiSEP GO ITD 02 MDA 2017). The authors also convey their sincere thanks to Prof. Perumana R. Sudhakaran, Department of Computational Biology and Bioinformatics, University of Kerala, Thiruvananthapuram, India, for his valuable suggestions and comments.
Authors’ Contributions
C.S.A. conceived the idea and designed and planned the experiments. A.M., S.C.R., and C.S.A. performed the data curation, analysis, and article drafting. P.R. helped with data analysis and making illustrations for the article. R.R., P.N., and T.S.K.P. reviewed and edited the article. All authors read and approved the final version of the article.
Author Disclosure Statement
The authors declare no conflicts of interest.
Funding Information
No funding was received for this article.