
Abstract
Background:
Orientia tsutsugamushi, causative agent of scrub typhus is an obligate intracellular parasite. We present information on isolation of this pathogen at a tertiary care centre in Vellore, Southern India.
Materials & Methods:
PBMCs (peripheral blood mononuclear cells) collected from suspected scrub typhus patients were inoculated into Vero and L929 cell lines and incubated at 37°C with 5% CO2 for 30 days. They were examined for presence of Orientia tsutsugamushi on 10, 15, 20 days post-inoculation and everyday thereafter for a maximum of 30 days post inoculation. The scrapings were subjected to Giemsa staining, IFA, 47kDa qPCR and transmission electron microscopy (TEM). The isolates were passaged 3–4 times to ensure viability and then stored in DMEM with 10% FBS (−80). Genotyping of the isolates was performed by amplifying a 650 bp segment of the TSA 56 (type specific antigen 56) gene.
Results:
Amongst the 50 samples inoculated, three were culture positive as confirmed by 47 kDa qPCR on 24th day post inoculation. This was further confirmed by Giemsa, IFA staining and TEM. The 650bp amplicons showed 99.5 to 100% homology with Orientia tsutsugamushi MW604716, MH003839, MW604718, MW604717, MH922787 and MH003838 strains. Phylogenetic analysis revealed that 2 isolates belong to TA763 genotype and one belongs to Gilliam genotype.
Conclusion:
Orientia tsutsugamushi has been isolated for the first time at Vellore, South India from PBMCs. Complete genomic analysis will give more information.
Introduction
Orientia tsutsugamushi is an obligate intracellular bacteria, the etiological agent of vector-borne zoonotic disease Scrub typhus (Watt and Parola, 2003). O. tsutsugamushi strains are classified into several subgroups based on the antigenic variations of the major surface protein called type-specific antigen 56 (TSA 56) (Kumar et al., 2019; Tilak and Kunte, 2019).
The infection is transmitted to humans (accidental dead-end host) by the bite of an infected larval stage of trombiculid mites commonly called as chiggers (Watt and Parola, 2003). In humans, O. tsutsugamushi invades the macrophages and vascular endothelial cells, escapes from the phagosomes, and propagates in the cytosol, inducing an acute febrile state. It often causes a deadly illness, if not treated with appropriate antibiotics, but the molecular pathogenicity is still poorly understood and no vaccine is currently available (Ko et al., 2013; Nakayama et al., 2008). In India, scrub typhus is a common cause of acute febrile illness and the estimated burden is 25.3% in such cases with an overall case fatality rate of 7% which increases to 40% in those with multiorgan dysfunction (Devasagayam et al., 2021).
The scrub typhus diagnosis is mostly based on serological techniques, and the serological reference standard test is the indirect immunofluorescence assay (IFA) (Blacksell et al., 2007). The scrub typhus IgM ELISA also has a similar sensitivity and specificity to the IFA (Blacksell et al., 2016). This assay utilizes the O. tsutsugamushi-specific recombinant 56-kDa antigen (Elangovan et al., 2019). PCR, especially real-time PCR (qPCR), is most useful for diagnosis in the early stage of scrub typhus infection. The common targets of these assays are the genes encoding the 56-kDa antigen and 47-kDa surface antigen, 16S rRNA, and groEL genes (Kim et al., 2011; Long et al., 2020; Masakhwe et al., 2018; Sonthayanon et al., 2006).
Isolation of O. tsutsugamushi in cell culture, though technically demanding, is useful for understanding the biology including studying the genomics and determining antimicrobial susceptibility (Luksameetanasan et al., 2007). Vero and L929 cell lines have been traditionally used to isolate and propagate O. tsutsugamushi (Ming et al., 2020; Tay et al., 2003). Growth of the organism is slow (20–30 days), therefore the results are of little use to the patient as well as the treating clinician (Ming et al., 2020; Vincent, 2016). However, it is important to obtain isolates of this bacteria to study the biology of the organisms including pathogenesis, virulence factors, host cell interactions, and genomic features. Further the availability of isolates will be the source of nucleic acids which can be used as positive control for various PCR assays, whole genome sequencing (Ming et al., 2020; Prakash et al., 2022; Tay et al., 2003). The culture isolates can also be useful to develop newer diagnostics and vaccines, which are region-specific (Ming et al., 2020).
Only few studies are available globally on isolation of O. tsutsugamushi and none from India (Prakash J.A., personal communication). We therefore proceeded to establish a standard protocol for isolation, characterization, and maintenance of O. tsutsugamushi from whole blood samples at our center.
Methodology
Sample collection and processing
Whole blood (8 mL) was collected in cell processing tubes (CPT) with sodium heparin (BD Vacutainer® CPT™, Franklin Lakes, NJ, USA) from clinically suspected scrub typhus patients. The collected samples were centrifuged at 1600 RCF for 20 min at room temperature. The separated plasma was aliquoted and stored; the monocyte layer was pipetted carefully and aliquoted into two tubes with the addition of equal volume of MEM/DMEM. One aliquot of monocytes was immediately transported to cell culture laboratory and inoculated into cell culture; other was used for DNA extraction using the Promega Wizard® Genomic DNA Purification kit (Promega Corporation, Madison, WI, USA) as per the manufacturer’s protocol. The extracted DNA was quantified using Nanodrop Spectrophotometer (ThermoFisher Scientific, Wxaltham, MA, USA) and stored at −80°C for molecular assays.
Isolation of O. tsutsugamushi in cell culture
The Vero and L929 cell line were maintained according to the protocol described by National Center for Cell Science (NCCS), Pune. The cells were grown in 25 cm2 flasks (Greiner) with growth medium (minimal essential medium [MEM]), Himedia, Thane, Maharashtra, India] with 10% fetal bovine serum (FBS, Merck, Rahway, NJ, USA) at 37°C and in a humidified atmosphere containing 5% CO2. At 80–100% confluence the cells were subcultured into a new flask (1:5) and maintained.
The peripheral blood mononuclear cells (PBMCs) with MEM were directly inoculated into a 10 cm2 Vero and L929 cell line at 80% confluence. The inoculated flasks were kept in a shaker for 30 min, then at 37°C for 2 h to facilitate infection. The inoculum was removed and replaced with fresh MEM with 2% FBS and incubated in a 5% CO2 incubator. Due to rapid acidification of MEM (Low glucose), the inoculated cell line was subjected to daily media change. To avoid frequent media change we switched to DMEM (with high glucose). For every 3 days once the spent medium was removed and replaced with fresh DMEM contained 2% FBS. The inoculated cell line were observed for morphological changes such as rounding of cells, floating cells, clumping of cells (Tay et al., 2003). The monolayer was checked by 47 kDa qPCR on the 10th day, 15th day, 20th day, and every day thereafter for a maximum of 30 days postinoculation. After 30 days of negativity the flasks were terminated whereas the positive culture was harvested and subcultured into 25 cm2 culture flask contained fresh Vero/L929 cell line and maintained in the same way. Giemsa staining, IFA (immunofluorescence assay) and transmission electron microscopy (TEM) were performed on the O. tsutsugamushi positive cultures (isolates).
Bacterial propagation and cryopreservation
The spent medium was removed from infected flasks and replaced with 5 mL of fresh DMEM. The infected cell lines were harvested by scraping using a sterile scraper. The detached cells were mixed well and subcultured onto fresh Vero cell line at a ratio of 1:5 (1 mL inoculum + 4 mL DMEM with 2% FBS per flask) and assessed for growth.
For cryopreservation, the infected cells were harvested with 5 mL of DMEM with 10% FBS and aliquoted into cryovials (1 mL). The vials were placed in isopropanol bath (Mr. Frosty freezing container, ThermoFisher Scientific, Waltham, MA, USA) and kept at −80°C for gradual freezing. After 24 h, the vials were transferred into liquid nitrogen for long-term preservation.
Giemsa staining
The infected cell line was pelleted by centrifugation at 4000 rpm for 20 min. The supernatant was discarded. A loop full of pellet was transferred to sterile glass slide and smear was made. The smear was fixed using methanol and air dried. The air-dried smear was flooded with Giemsa Stain, modified solution (Merck, St. Louis, MO, United States) for 20 min. The slide was washed with distilled water and observed under light microscope (100×).
Immunofluorescence assay
Slides were prepared using the abovementioned method. The smear was flooded with primary antibody (IgM ELISA positive serum with OD > 2.00) and incubated for 30 min at room temperature. The unbound antibody was washed four times using wash buffer and air dried. Antihuman FITC (Dako, Glostrup, Denmark)-labeled antibody (IgM) was added to the air dried smear and incubated at room temperature for 30 min. The unbound conjugate was washed four times using wash buffer and air dried. The slide was mounted and observed under fluorescent microscope.
Transmission electron microscopy
The cell culture scrapings were subjected to centrifugation at 4000 rpm for 20 min. The pellet was prefixed with 3% glutaraldehyde and washed in buffer. Then it was postfixed by 1% osmium tetroxide followed by washing. Then the pellet was blocked with 2% agarose. The block was subjected to dehydration using ascending series graded alcohol (50%–100%), followed by infiltration by propylene oxide and epoxy resin. Finally, embedded in siliconized rubber mold with epoxy resin and kept in incubator at 60°C for 48 h for polymerization. The blocks were subjected to thick section to select the area of interest then subjected to ultrathin section (below 100 nm). The ultra-thin section was taken in copper grid and stained using uranyl acetate and Reynold’s solution which gives contrast. The sections are observed under TEM (FEI Philips Technai T12 Spirit, FEI, Hillsboro, OR, USA) and photographed.
Extraction of DNA from culture isolate
The DNA was extracted from infected cell lines using the Promega Wizard® Genomic DNA Purification kit (Promega Corporation, Madison, WI, USA) as per the manufacturer’s protocol. The extracted DNA was quantified using Nanodrop spectrophotometer (ThermoFisher Scientific, Waltham, MA, USA) and stored at −80°C for further use.
Real-time PCR
The extracted DNA was subjected to real-time PCR targeting 47 kDa gene of O. tsutsugamushi on an Applied Biosystems™ 7500 Real-Time PCR System (ThermoFisher Scientific, MA, USA). The amplification was performed using TaqMan Fast Advanced Master mix (ThermoFisher Scientific, Waltham, MA, USA). Each reaction mixture (25 µL) contained 5 µL of template DNA, 10 pmol of each primer, 12.5 µL of Master Mix, and 5 pmol of the probe. The PCR conditions included an initial denaturation at 95°C for 5 min followed by 40 cycles of 95°C for 30 s and 60°C for 1 min. The Ct value ≤35 was considered as positive (Prakash et al., 2022).
Genotyping of culture isolate
The O. tsutsugamushi DNA was subjected to a nested PCR protocol as described by Horinouchi. This amplifies a 650 bp segment of the TSA 56 gene (includes VD-1 to VD-III) and was performed as described previously (Kumaraswamy et al., 2024; Masakhwe et al., 2018). The procedure is briefly given below.
The first round of PCR was performed with 12.5 µL of mastermix, 10 pm of outer primers RTS 8, and RTS 9 with 5 µL DNA template. The amplicons were generated using the following reaction conditions: 95° C for 5 min; 35 cycles of 94° C for 50 s, 57° C for 60 s, 72° C for 1.30 min, and final extension of 72° C for 7 min. The second round of PCR was performed using inner primers RTS 6 and RTS 7 with 2 µL of first round product as DNA template. The cycling conditions are as same as first round except the extension time was reduced to 1 min. The PCR product was analyzed using agarose gel electrophoresis system and visualized by gel documentation (Gel Doc, Bio-Rad, Hercules, California, USA). The 650 bp positive fragment was purified by Wizard® SV Gel and PCR Clean-Up System (Promega Corporation, Madison, WI, USA). The amplified fragment was subjected to preclean up using Exosap-IT Applied Biosystems (ThermoFisher Scientific, Waltham, MA, USA) method with reaction conditions as follows: 37°C for 15 min, 80°C for 15 min, and 15°C for 2 min. The preclean up product was used as template for sequencing PCR to amplify individual strands of the fragments BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA). The amplified segment was subjected to post clean-up using HighPrep™ DTR Clean-up System (MagBio Genomics Inc., Gaithersburg, MD, USA) and eluted with molecular grade water. The eluted product was loaded in Genetic Analyzer 3500 (ThermoFisher Scientific, Waltham, MA, USA) and the raw reads were analyzed using the Bio-Edit software.
Phylogenetic tree construction
The obtained sequence was subjected to BLAST analysis. Alignment was performed using Clustal Omega (Sievers et al., 2011) with 36 reference sequences retrieved from GenBank-NCBI and the phylogenetic tree was established using IQTREE software: A fast and effective stochastic algorithm was used for estimating maximum likelihood phylogenies (Nguyen et al., 2015).
Results
Of the 50 samples inoculated into cell culture, O. tsutsugamushi was isolated in Vero cells from 3 samples (confirmed by 47 kDa qPCR) after 24th day of inoculation (refer to Table 1). The positive samples (Vellore isolate 1–3) were subcultured into 25cm2 flask (Fig 1). The three isolates were genotyped by using the 56 kDa type-specific antigen partial gene sequence (650 bp). BLAST analysis of this partial sequence showed 99.5%–100% homology with MW604716, MH003839, MW604718, MW604717, MH922787, and MH003838 O. tsutsugamushi strains. Phylogenetic analysis revealed that the Vellore isolates 1 and 2 belong to TA763 whereas the Vellore isolate 3 belongs to Gilliam genotype FIG. 5.).
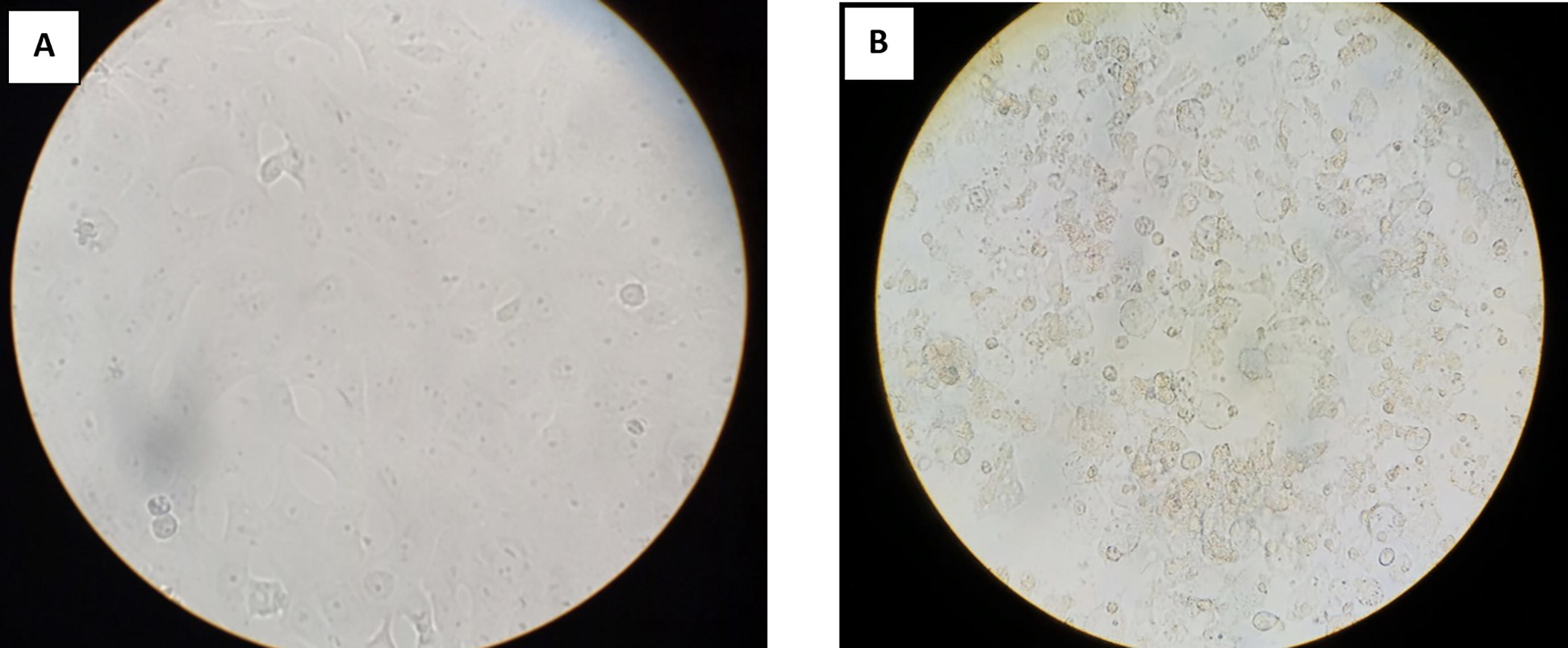
Culture Isolates Positive for 47 kDa qPCR
Undetected.
Among the three, two isolates were cryopreserved and one isolate was maintained for further morphological analysis such as Giemsa, IFA, and TEM.
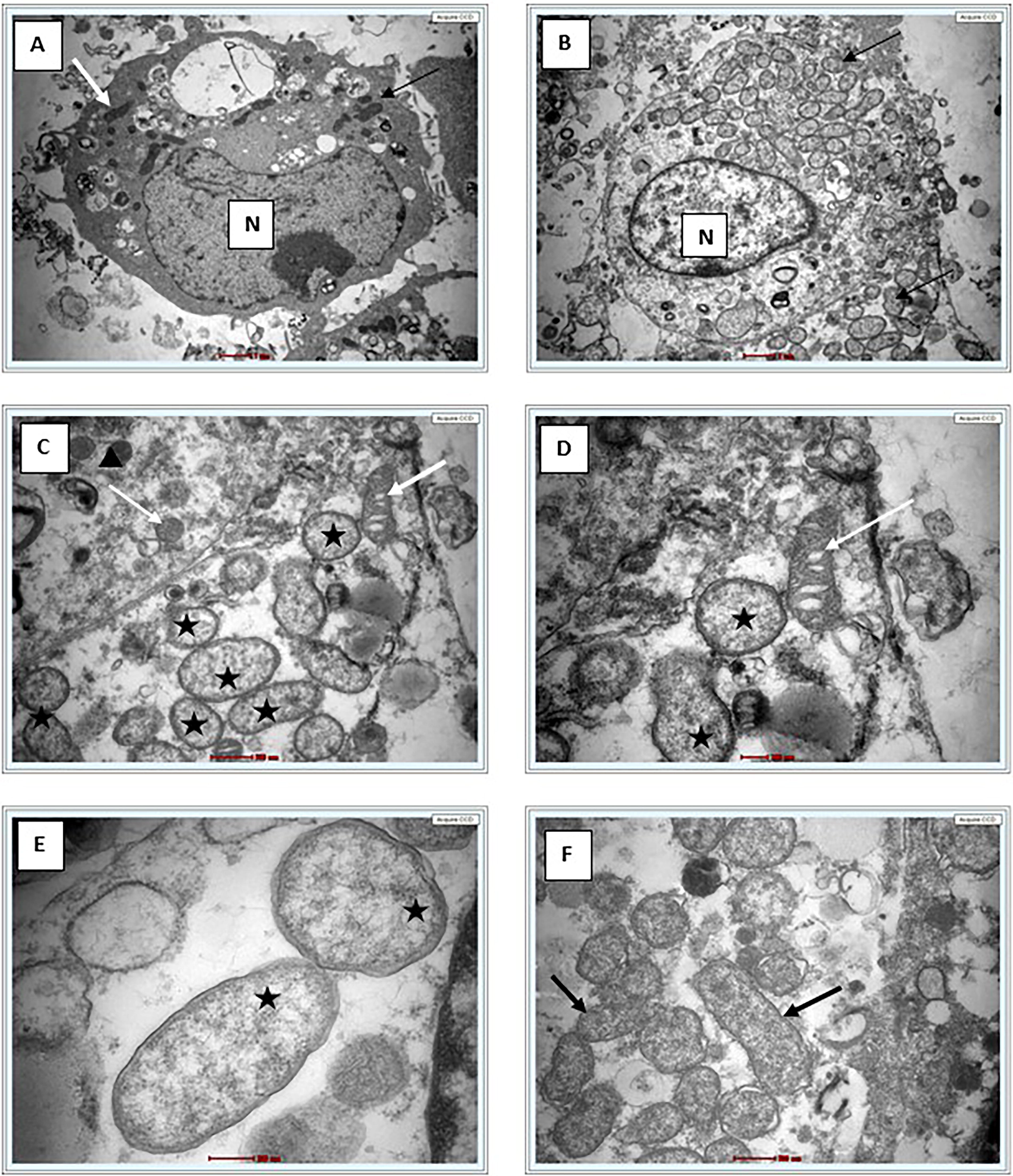
Giemsa staining revealed purple color small rod-shaped organisms in Vero cells (Figure 2). IFA showed bright green stained organisms as shown in Figure 3. Further, we analyzed the ultrastructural morphology of O. tsutsugamushi in infected Vero cell by TEM. TEM showed O. tsutsugamushi in infected Vero cell cytoplasm, the bacterium was round-to-oval shaped with characteristic double membrane (Fig. 4B and E: indicated by black star), whereas the mitochondria are smaller, darker, spherical/elliptical with cristae seen in higher magnification (Fig. 4C and D: white arrow head). Extracellular O. tsutsugamushi are shown in Figure 4F (black arrows).
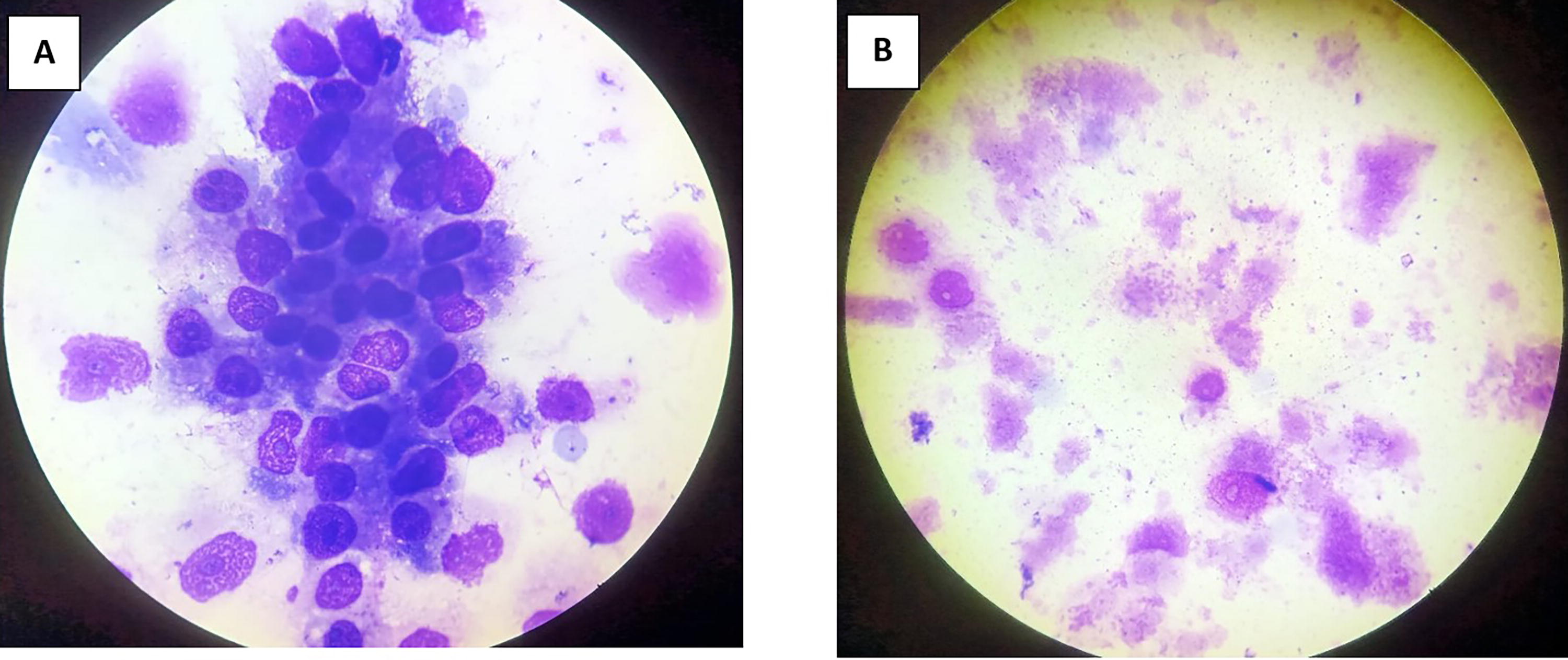
Giemsa staining of Vellore isolate 1 isolate.

Indirect Fluorescence Assay of Vellore isolate 1 isolate.
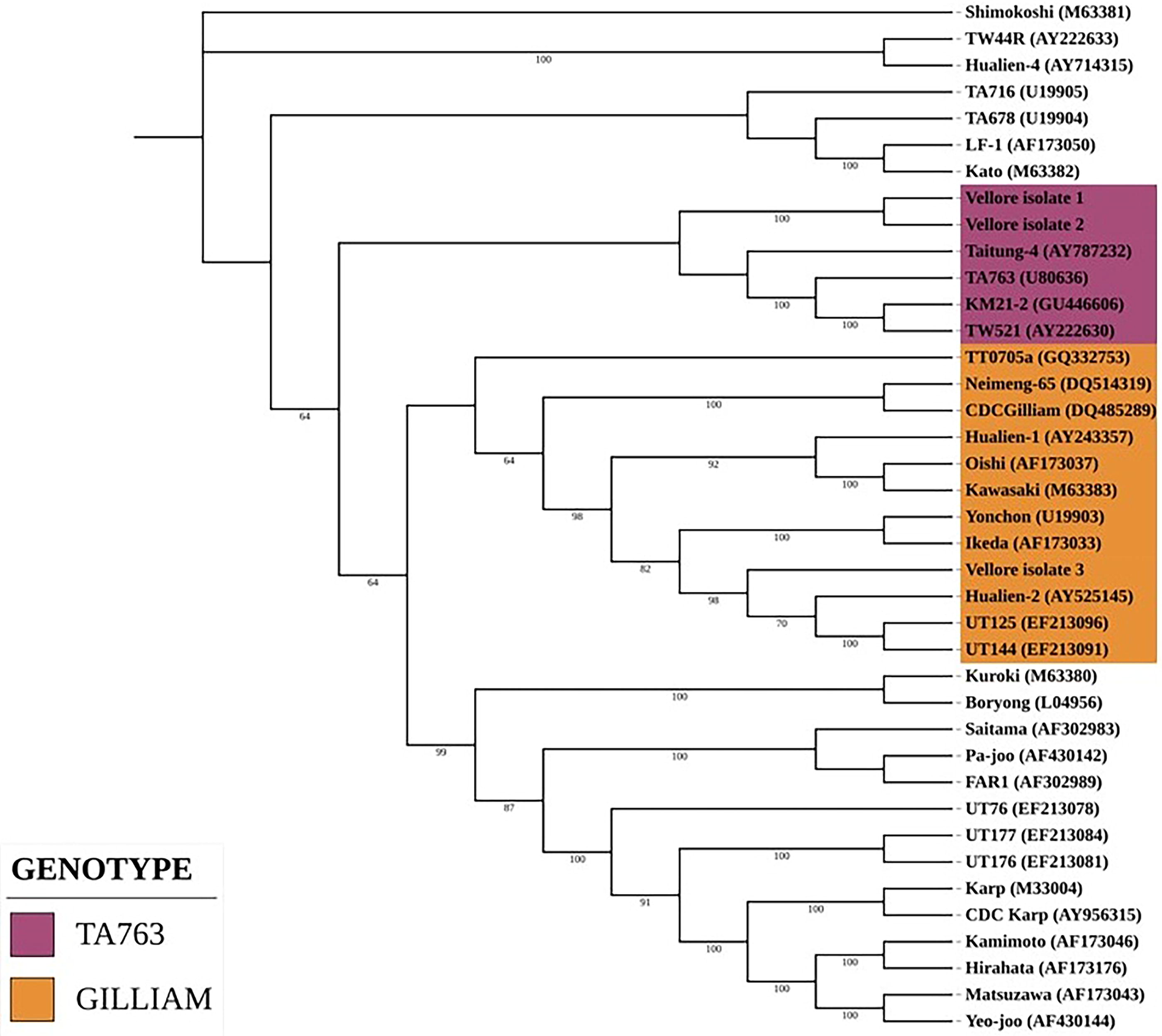
Phylogenetic analysis of Vellore isolates.
Discussion
The in vitro isolation of O. tsutsugamushi from clinical samples is important to understand the genomic diversity of the pathogen for development of vaccines and diagnostics. There are few reports of successful isolation of O. tsutsugamushi in the recent past (Luksameetanasan et al., 2007; Ming et al., 2020) and most laboratories capable of isolating this agent use both Vero and L929 cells (Prakash JAJ, Personal communication). Ming et al. reported isolation of O. tsutsugamushi in 231 (7.16%) samples of the 3227 tested over a 6-year period (2008–2014) using Vero and or L929 cells. All these were confirmed by scrub typhus IFA & 47 kDa qPCR. Genotyping data has not been provided for these 231 isolates (Ming et al., 2020). Luksameetanasan R. et al. isolated O. tsutsugamushi in 12 (46.15%) out of 26 buffy coat samples over a 6-month period (June–November 2004) (Luksameetanasan et al., 2007). Nine of these isolates belonged to Karp genotype, two to Gilliam, and one belonged to TA763. In this study, we have successfully isolated the O. tsutsugamushi from whole blood sample. The use of 10cm2 flask for initial isolation is a simplified method to get an isolate from minimal amount of sample. The 10 cm2 culture tube has a wide mouth opening for easy manipulation to sample the infected cell line/supernatant. Initially, MEM was used for isolation but MEM depleted faster, this required frequent media change. Hence, we switched to DMEM which is more suitable for isolation and maintenance of O. tsutsugamushi as it has longer incubation period. DMEM is complex media contains four-fold increased concentrations of amino acids and vitamins. Cells grow faster and deplete the nutrients slower in DMEM.
Phylogenetic analysis of the partial gene sequence (Horinouchi protocol) which covers the variable domain I, II, and III revealed our isolates belongs to TA763 and Gilliam genotype (Kumaraswamy et al., 2024; Masakhwe et al., 2018). One isolate was morphologically characterised by Giemsa staining, IFA and Electron microscopy. The size and shape of the bacterium resembles mitochondria, the cristae and the difference in electron density helped to identify the O. tsutsugamushi (Fig. 4C and D) as described by Paris and coworkers and cited in the literature by Gemma Vincent (Prakash et al., 2022).
Conclusion
We have successfully isolated and characterized O. tsutsugamushi for the first time at our center and most likely in India from PBMCs. Based on the partial TSA56 gene sequence our isolates belong to TA763 and Gilliam genotype. Complete 56 kDa and whole genome analysis is needed to definitively identify the genotype. More number of samples from suspected cases is being processed for isolation in cell culture to obtain information regarding the genotypes of O. tsutsugamushi circulating in and around Vellore.
Footnotes
Authors’ Contributions
J.K., K.G., A.K., S.D., A.K.P.P., and J.A.J.P.: Conceptualization and methodology. J.K., A.K., S.D., K.G., A.K.P.P., and J.A.J.P.: Investigations and data acquisition. J.K., A.K., and J.A.J.P.: Analysis and data interpretation. J.K., A.K., and S.D.: Drafting the article. K.G., A.K.P.P., and J.A.J.P.: Critical revision of the article. J.A.J.P.: Supervision and funding acquisition.
Author Disclosure Statement
The authors declare that they have no conflicts or competing interests to declare.
Funding Information
The study was funded by
Ethics Approval
The study was approved by Institution Review Board (Silver, Research and Ethics Committee) of the Christian Medical College (CMC), Vellore, India. IRB Min. No. 11942, Dated 27.03.2019.