
Abstract
Intracranial infection of mice with lymphocytic choriomeningitis virus (LCMV) results in a lethal neurological disease termed lymphocytic choriomeningitis (LCM) that is mediated by antiviral CD8+ T cells. Previous studies have implicated the chemokine receptor CXCR3 and its ligand CXCL10 in CD8+ T cell trafficking in the brain and in the lethal disease following intracranial infection of mice with the LCMV-Traub strain. Here we investigated the role of CXCR3 in LCM following intracranial infection of mice with the LCMV-Armstrong strain. Significant induction of both CXCL9 and CXCL10 RNA and protein was seen in the central nervous system (CNS) in LCM. Cellular localization of the CXCL9 and CXCL10 RNA transcripts was identified predominantly in infiltrating mononuclear cells, as well as in subpial and paraventricular microglia (CXCL9) and astrocytes (CXCL10). Despite a primary role of interferon (IFN)-γ in inducing the expression of the CXCL9 gene, and to a lesser extent the CXCL10 gene in LCM, the absence of the IFN-γ receptor did not influence the disease outcome. This finding suggested that these chemokines may not play a major role in the pathogenesis of LCM. To evaluate this possibility further the development of LCM was examined in mice that were deficient for CXCR3. Surprisingly, in the absence of CXCR3 there was no alteration in mortality, cytokine expression, or T cell infiltration in the CNS, demonstrating that in contrast to LCMV-Traub, CXCR3 is not involved in the pathogenesis of LCMV-Armstrong–induced neurological disease in mice. Our findings indicate that despite similar immunopathogenetic mechanisms involving antiviral CD8+ T cells, whether or not CXCR3 signaling has a role in LCM is dependent upon the infecting strain of LCMV.
Introduction
The directed trafficking of T cells during inflammation is mediated by a group of cytokines termed chemokines that interact with specific cell surface receptors (8,39). In particular, the chemokine receptor CXCR3 is involved with the trafficking of activated T cells in a number of different immune-mediated pathologies (reviewed in 22). CXCR3 is located mainly on activated CD4+ Th1-type and CD8+ T cells (20,25,37). CXCR3 binds the structurally related CXC family chemokines CXCL9 (MIG), CXCL10 (IP-10), and CXCL11 (ITAC) (12,25,26). The Cxcl11 gene contains a frameshift mutation in C57BL/6 mice that is predicted to result in a nonfunctional protein (see Genbank accession numbers NT 109320, NT 039339, and NW 001030791). CXCL9, CXCL10, and CXCL11 lack the Glu-Leu-Arg (ELR) motif common to other members of the CXC chemokine group, and are all inducible by interferon-γ (IFN-γ) (12,15,27,38). While these chemokines are not detectable in the healthy brain, their expression is highly upregulated following infection of the CNS with various viruses (2,3,10,21,23,24). Deficiency of CXCR3 has been associated with reduced trafficking to and/or positioning of T cells in the CNS in a number of different virally-induced disease models, such as West Nile virus encephalitis (45), mouse hepatitis virus encephalitis (43), and dengue virus encephalitis (18).
The potential involvement of the CXCR3/CXCL9-CXCL10 chemokine axis in the development of LCM is supported by several studies. Following IC LCMV infection in mice, CXCL10 RNA levels are induced in the CNS prior to leukocyte recruitment (2,3,9). Furthermore, CXCR3-deficient mice (10) and CXCL10-deficient mice (9) were reported to be partially protected from lethal LCM, and increased survival in these mice was paralleled by reduced infiltration of the CNS parenchyma by T cells, and reduced levels of IFN-γ in the CNS (10). Together these studies suggested that the intra-parenchymal positioning of antiviral CD8+ T cells in the brain following IC LCMV infection is mediated by the interplay of CXCL10 produced in the brain with its receptor CXCR3 on antiviral CD8− T cells.
The aim of our study was to determine further the cellular localization and role of IFN-γ in regulating CXCL9 and CXL10 gene expression in LCM. Moreover, the finding here that mice lacking IFN-γ receptor signaling showed unaltered susceptibility to LCM despite compromised induction of CXCL9 and CXCL10, led us to reassess the role of CXCR3 signaling in LCMV-induced immune pathology and lethal disease. In contrast to previous studies (9,10), we observed that deficiency in CXCR3 signaling had no effect on the development of LCM. In summary, our findings demonstrated that CXCR3 is dispensable for LCM in mice infected intracranially with LCMV-Armstrong.
Materials and Methods
Animals
The generation and characterization of the CXCR3−/− mice was described previously (16). The mice used in the present study were backcrossed for 12 generations to the C57BL/6J strain. Wild-type C57BL/6 mice were purchased from the Animal Resources Centre (Canning Vale, WA, Australia) and served as controls. The animals were kept under pathogen-free conditions in the Blackburn animal facility of the University of Sydney. Ethical approval for the use of all mice in this study was obtained from the University of Sydney Animal Care and Ethics Committee.
LCMV infection
Adult mice between 3 and 6 mo of age were anesthetized and injected IC into the frontal cortex with 200 pfu Armstrong 53b strain of LCMV in 20 μL vehicle (PBS + 10% fetal bovine serum), or with 20 μL vehicle alone (sham). For survival studies five or six mice per group were used.
Tissue processing for histology, protein, and RNA isolation
At day 6 following infection mice were euthanized and their brains removed for immunohistochemistry, dual-label in situ hybridization (ISH), and RNase protection assay (RPA). For immunohistochemistry, tissue was embedded without prior fixation in Tissue Tek (Sakura Finetek, Zoeter-woude, Netherlands) and flash frozen in liquid nitrogen–cooled isopentane. For dual-label ISH, brains were removed and placed immediately in PBS-buffered 4% paraformaldehyde for 24 h at 4°C and were subsequently embedded in paraffin. For protein and RNA isolation tissue was immediately flash frozen in liquid nitrogen and stored at −80°C pending isolation of RNA or protein.
Immunohistochemistry
For immunohistochemistry 12-μm-thick frozen sections were fixed in ice-cold methanol:acetone (50:50) for 45 sec. Primary antibodies (CD4 and CD8, 1:200; BD Pharmingen, Ryde, Australia) were incubated overnight at 4°C. After washing in PBS, a biotinylated secondary antibody (1:200, 45 min; Vector Laboratories, Burlingame, CA) and horseradish peroxidase–coupled streptavidin (1:200, 30 min; Vector Laboratories) was used. Diaminobenzidine/H2O2 reagent (Vector Laboratories) was applied as the immunoperoxidase substrate according to the manufacturer's instructions. Sections were counterstained for 15 sec with hematoxylin (Sigma-Aldrich, St. Louis, MO) prior to dehydration and mounting of cover slips. Immunohistochemical stained sections were examined under a DM4000B bright field microscope (Leica, Wetzlar, Germany) and images were taken using a Spot Flex camera and Spot V4.5 software (Diagnostic Instruments, Sterling Heights, MI).
Protein isolation and ELISA
For protein isolation, the brain was homogenized in PBS containing protease inhibitors (Merck, Kilsyth, Vic, Australia) and the protein concentration was determined using a commercially available Bradford assay (Bio-Rad, Regents Park, New South Wales, Australia). The specific levels of CXCL9 or CXCL10 protein in the brain homogenates was determined by enzyme-linked immunosorbent assay (ELISA) using commercially available kits (R&D Systems, Minneapolis, MN). For each assay a standard curve was generated with the limit of detection for CXCL9 and CXCL10 being 3 pg/mL and 2 pg/mL, respectively. All samples were measured in duplicate.
RNA isolation and RNAse protection assays
Total RNA was isolated using TriReagent (Sigma-Aldrich) according to the manufacturer's instructions. RNase protection assays (RPAs) for the detection of cytokine RNAs were performed as described previously (33). RNA samples were hybridized with labeled cytokine, chemokine, or LCMV-NP probes. The probes for CXCL9, CXCL10, CXCR3 (2,4), IFN-γ, TNF, IL-1α, IL-1β, IL-6 (2,3), and LCMV-NP (41) were described previously. The genomic clone RPL32-4A (L32; kindly provided by M. Hobbs, The Scripps Research Institute, San Diego, CA) served as a probe for the ribosomal protein L32. This was included as an internal control for RNA loading during RPA analysis.
Dual-label in situ hybridization and immunohistochemistry
For ISH 5-μm-thick paraffin-embedded sections were incubated with P33-labelled cRNA probes transcribed from linearized plasmid constructs containing the CXCL9, CXCL10, and LCMV-NP cDNA inserts, and processed for in situ hybridization histochemistry as described previously (3,40). Sections for immunohistochemistry were reacted with antibodies to detect astrocytes (rabbit anti-glial fibrillary acidic protein, 1:500; DAKO Cytomation, Botany, Australia), and T lymphocytes (rabbit anti-CD3, 1:200; DAKO Cytomation). Microglia were detected with biotinylated lectin from Lycopersicon esculentum (Sigma-Aldrich, 1:50). Bound antibody or lectin was detected using Vectastain ABC kits (Vector Laboratories), and diaminobenzidine/H2O2 reagent (Vector Laboratories) as the immunoperoxidase substrate.
Statistical analysis
For the quantification of RPA autoradiography, densitometric analysis of each band was performed using NIH Image J software. The level of the individual RNA or protein density were normalized to that of the loading control L32 and the mean ± SEM calculated using Prism software (GraphPad Inc., San Diego, CA). Statistical significance was calculated using one-way ANOVA or Student's t-test where appropriate, with p < 0.05 considered to be significant.
Results
CXCL9 and CXCL10 RNA and protein levels were upregulated in the CNS following LCMV infection
Following IC infection with LCMV-Armstrong, wild-type C57BL/6 mice (WT mice) developed lethal LCM between 6 and 8 d postinfection (Fig. 1A). LCM is known to be caused by antiviral CD8+ T cells that are recruited to the brain (6,13). These T cells express the chemokine receptor CXCR3 and expression of the CXCR3 ligand, CXCL10, in the CNS of LCMV-Armstrong-infected mice has been shown previously (2,3). To confirm these findings and determine whether the gene for Cxcl9 was also induced, RPA was performed. The mRNA transcripts corresponding to CXCL9 and CXCL10 were not detectable in the CNS in non-infected (data not shown) or sham-infected WT mice (Fig. 1B and C). In contrast, both CXCL9 and CXCL10 mRNA transcripts were present at high levels in the CNS at day six following LCMV infection. The level of CXCL10 mRNA induced in the brain by LCMV infection was higher than the corresponding level of CXCL9 mRNA. Both CXCL9 and CXCL10 protein were also induced in the brain following LCMV infection, and paralleled the RNA levels, with CXCL10 being markedly higher than CXCL9 (Fig. 1D). In summary, these findings showed that there is significant production of both CXCL9 and CXCL10 in the brains of mice with LCM.
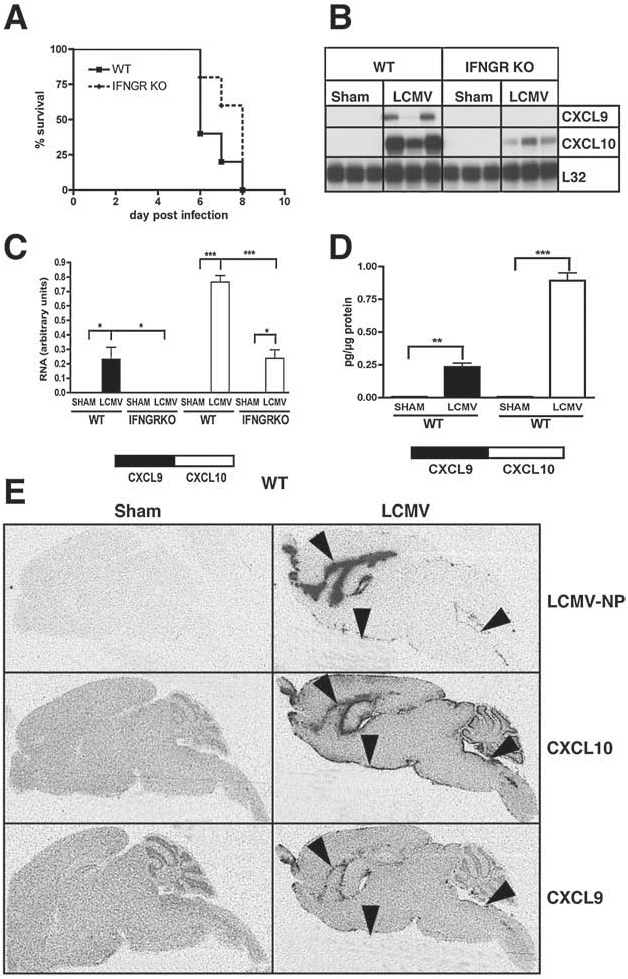
Cerebral Cxcl9 and Cxcl10 gene expression in LCM and the effect of IFN-γ. WT or IFN-γ-receptor (IFNGR) signalling knockout (KO) mice were injected IC with LCMV-Armstrong (200 pfu) or vehicle alone (sham). (
CXCL9 and CXCL10 RNA co-localized with LCMV but showed differential cellular localization in the CNS of LCMV-infected mice
We next examined the anatomical and cellular expression of the Cxcl9 and Cxcl10 genes in LCMV-infected WT mice by dual-label ISH. ISH revealed that RNA for CXCL9 and CXCL10 was confined largely to the meninges, ventricular ependyma, and choroid plexus, and overlapped with RNA for the LCMV nucleoprotein (LCMV-NP) (Fig. 1E; arrowheads). The expression of both genes in the meninges (Fig. 2A–D; arrowheads) and choroid plexus (data not shown) was confirmed by microscopic examination. In the subpial and periventricular parenchyma CXCL9 RNA also co-localized with lectin-positive macrophages/microglia (Fig. 2C; white arrows) and was not detected in GFAP-positive astrocytes (Fig. 2A; black arrow). In contrast to this, CXCL10 RNA was found predominantly in subpial astrocytes (Fig. 2B; white arrows), but was largely absent in macrophages/microglia (Fig. 2D; black arrows). Additionally, high levels of CXCL9 (Fig. 2E) and CXCL10 RNA (Fig. 2F) were identified in infiltrating mononuclear cells. Some CD3+ T cells present in the mononuclear cell infiltrates were positive for both CXCL9 (Fig. 2E; white arrows) and CXCL10 (Fig. 2F; white arrows) RNA.

Cellular localization of CXCL9 and CXCL10 RNA in the brain in LCM. WT mice were infected IC with LCMV-Armstrong (200 pfu) and the brains were removed at day 6 postinfection. ISH for CXCL9 (
Deficiency of the IFN-γ receptor resulted in the absence of Cxcl9 and reduced Cxcl10 gene expression, but did not alter clinical LCM
IFN-γ is known to be an important inducer of the Cxcl9 and Cxcl10 genes. Therefore, we next investigated the cerebral expression of the genes for Cxcl9 and Cxcl10 in LCM in mice that lacked IFN-γ-receptor signaling (IFNGR knockout [KO] mice). Like WT mice, sham-infected IFNGR KO mice showed no detectable CXCL9 or CXCL10 mRNA. Furthermore, in IFNGR KO mice CXCL9 mRNA remained undetectable in the CNS at day six following LCMV infection, while CXCL10 RNA levels were significantly reduced compared with LCMV-infected WT mice (Fig. 1B and C). Despite the marked decrease in the level of expression of the Cxcl9 and Cxcl10 genes, deficiency of the IFN-γ receptor had no impact on the survival or the clinical symptoms of the LCMV-infected mice (Fig. 1A). These findings indicated that IFN-γ-receptor signaling was not critical for the development of LCM.
Deficiency of CXCR3 did not alter the clinical course of lethal LCM nor the levels of LCMV-NP RNA in the brain
Previous studies have reported that deficiency of CXCR3 partially protected mice from the LCMV-induced lethal disease and linked CXCR3 signaling to parenchymal infiltration of CD8+ T cells (10). However, our findings above in the IFN-γ-receptor-deficient mice were at odds with this finding. Therefore, here we re-examined the role of CXCR3 in the development of LCM induced by LCMV-Armstrong. Following IC infection we did not observe any significant difference in the disease outcome in CXCR3 KO mice compared with similarly infected WT controls (Fig. 3A). Independent of the genotype, all mice died between 7 and 9 d postinfection. There were no observable differences in the clinical symptoms and both WT and CXCR3 KO mice showed characteristic cerebral seizures of similar severity. Furthermore, LCMV-NP RNA levels as determined by RPA were similar in infected WT and CXCR3-deficient mice (Fig. 3B and C). These findings clearly indicated that CXCR3 signaling is dispensable for the normal development of LCM.
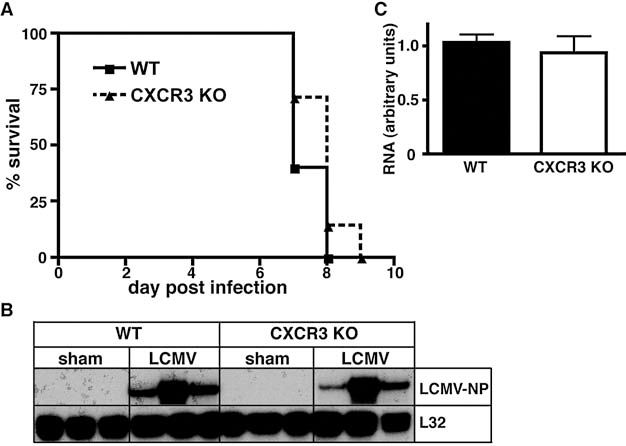
Survival analysis and brain viral transcript levels in WT and CXCR3 KO mice with LCM. WT or CXCR3 KO mice were infected IC with LCMV-Armstrong (200 pfu) or vehicle alone (sham) (
Deficiency of CXCR3 did not alter the levels of IFN-γ nor other cytokine RNA transcripts induced in the CNS
To determine if the absence of CXCR3 signaling altered cytokine production following LCMV infection, we next examined by RPA the cerebral RNA levels of several cytokines (Fig. 4). RNA for IFN-γ (Fig. 4A and B), TNF, IL-1α, IL-1β, and IL-6 (Fig. 4C and 4D) was not detectable in the CNS of sham-infected WT and CXCR3 KO mice. By contrast, elevated RNA levels for all five cytokines were present in the brains of LCMV-infected WT and CXCR3 KO mice at day six postinfection. In line with the unaltered clinical disease and virus RNA levels, no significant differences were observed in the level of any of these cytokine RNA transcripts between infected WT and CXCR3 KO mice.

Cytokine RNA levels in WT and CXCR3 KO mice with LCM. WT or CXCR3 KO mice were infected IC with LCMV-Armstrong (200 pfu) or vehicle alone (sham) and the brains were removed at day 6 postinfection. (
The distribution of CD4+ and CD8+ T cells was not altered by CXCR3 deficiency in LCM
CXCR3 signaling has been linked to T cell recruitment and positioning in tissues in a number of cell-mediated immune diseases including LCMV infection (10). Here we investigated by immunohistochemistry the spatial distribution of CD4+ and CD8+ T cells in the brains of WT and CXCR3 KO mice with LCM. In sham-infected mice only very few CD4+ and CD8+ T cells were present, and they were most likely within vessels (data not shown). In contrast, in LCM, large numbers of both CD4+ (Fig. 5A and E) and CD8+ T cells (Fig. 5C and G) were present in the meninges, the ventricles, and the choroid plexus of WT mice. However, very few T cells were observed within the CNS parenchyma adjacent to the meninges (not shown) or the ventricles (Fig. 5). In LCMV-infected CXCR3 KO mice the numbers and distribution of CD4+ (Fig. 5B and F) and CD8+ T cells (Fig. 5D and H) did not change when compared with LCMV-infected WT mice. In summary, these findings indicated that the absence of CXCR3 signaling did not significantly alter the trafficking to or positioning of T cells in the brain during LCM.

Localization of CD4+ and CD8+ T cells in WT and CXR3 KO mice with LCM. WT or CXCR3 KO mice were infected IC with LCMV-Armstrong (200 pfu) or vehicle alone (sham) and the brains were removed at day 6 postinfection. Immunohistochemistry for CD4 (
Discussion
The lethal neurological disease that follows intracranial infection of immunocompetent mice with LCMV is mediated by antiviral CD8+ T cells that are recruited to the infected brain and accumulate within the meninges and choroid plexus (1,11,42). However, the precise mechanisms that ensue and lead to the death of the animals are currently unknown. Since a key step in the pathogenesis of this disorder is the trafficking of antiviral CD8+ T cells to the CNS, there has been considerable focus on the potential involvement of chemokines in this process (2,3,28,29). Here we found that intracranial infection with LCMV induced high levels of CXCL9 and CXCL10 RNA and protein in the CNS. Expression of the gene for CXCL9 was closely linked temporally and spatially to the infiltrating mononuclear cell population, indicating that this chemokine is unlikely to be involved in the initial recruitment of these cells to the CNS in LCM. In a previous study, expression of the Cxcl10 gene was induced early after infection with LCMV and prior to the recruitment of mononuclear cells (2). Moreover, in the current study CXCL10 RNA localization included mononuclear cells as well as astrocytes immediately adjacent to the meninges. The differences in the expression pattern of the Cxcl9 and Cxcl10 genes suggested that these chemokines may have distinct roles in LCM that could involve effects on different target cell populations.
Expression of both chemokine genes was either totally (Cxcl9) or partially (Cxcl10) dependent on IFN-γ-receptor signaling. The dependence of the induction of cerebral Cxcl9 gene expression on IFN-γ during LCM has not been described previously. IFN-γ is also the primary mediator of Cxcl9 gene expression in myelin oligodendrocyte glycoprotein–induced experimental autoimmune encephalomyelitis (MOG-EAE) (7). In contrast to the Cxcl9 gene, both IFN-γ-dependent and -independent pathways mediate expression of the Cxcl10 gene in LCM. Indeed, it has been shown previously that other cytokines such as type I interferons (IFNs) and TNF-α (30,31,35,36), as well as microbial products such as LPS and HIV gp120 (4,31), can induce the expression of the Cxcl10 gene in a variety of different cell types. These differences in the regulation of Cxcl9 and Cxcl10 gene expression can best be explained by differences in the promoters for both genes that contain distinct transcription factor binding sites (17,32,44).
The high levels of CXCL9 and CXCL10 RNA and protein in the CNS of LCMV-infected mice and their distinct cellular expression patterns are consistent with the idea that these chemokines may play a role in the pathogenesis of LCM. However, despite the absence of CXCL9 RNA and significantly reduced CXCL10 RNA levels in the CNS, LCMV-infected mice lacking IFN-γ signaling did not exhibit any change in the clinical course of the disease. This finding is consistent with those of previous studies that found no requirement for IFN-γ in the development of LCM (28,34), and led us to question whether there was a role for CXCL9 and CXCL10 in this virally-induced immune-mediated disease process. Nevertheless, it could be argued that the low levels of CXCL10 RNA found in the CNS of the IFNGR KO mice may have provided sufficient signaling to contribute to LCM. Indeed, there is evidence that CXCL10 but not CXCL9 has a role in this model. Thus, mice with CXCL10 deficiency were reported to be partially protected from lethal intracranial LCMV infection (9).
In view of the possibility that low levels of CXCL10 in IFN-γ-receptor-deficient mice might be sufficient to elicit a meaningful biological effect in LCM, we investigated this virally-induced immune-mediated disease process in mice that lacked CXCR3. A previous report had established that the absence of CXCR3 was associated with a significant increase in survival of mice infected intracranially with LCMV (10). However, we could not confirm this earlier report with our findings here. Thus, we observed that CXCR3-deficient mice had a similar time to disease onset and mortality rate as wild-type controls. In further support of the lack of a role for CXCR3 in LCM, the cerebral expression of the LCMV-NP RNA as a marker for viral load, and a number of cytokine genes, including IFN-γ, were similar in the CXCR3-deficient and wild-type mice. The results reported by Christensen and co-workers in CXCR3- and CXCL10-deficient (9,10) mice suggested that a primary role of this chemokine-signaling pathway in LCM was to position antiviral effector T cells in the parenchyma of the brain. How the positioning of CD8+ T cells within the parenchyma in LCM could account for the mortality of mice infected intracranially with LCMV is not clear. Our studies here revealed that compared with the meninges and choroid plexus there were relatively few CD8+ T cells in the sub-meningeal or paraventricular parenchymal regions of the brain in mice with LCM following LCMV-Armstrong infection. Thus it is highly unlikely that CXCR3 signaling is involved in parenchymal T cell accumulation and/or positioning and is the cause of mortality in LCM that was observed in our studies.
So how can these marked differences in findings be explained between the studies of Christensen and colleagues (9,10) and those reported here? In all three studies C57BL/6 mice were used, in which the route of infection (IC) was the same and the doses of virus were similar. However, while Christensen and co-workers used the LCMV-Traub strain, our studies here used the LCMV-Armstrong strain. Both strains show marked genetic variation, resulting in different biological behavior such as survival following peripheral infection (14). However, both LCMV strains cause a lethal disease with a similar time-course following IC inoculation in WT mice, and to the best of our knowledge, no differences in cell tropism and virus spread following IC infection with either LCMV-Armstrong or LCMV-Traub have been reported. Yet, subtle differences in viral spread and the dynamics of the antiviral response make it at least conceivable that CXCR3 has a greater impact on neurological disease induced by LCMV-Traub versus LCMV-Armstrong. It is of note that the penetrance of the protective phenotype was not complete in the LCMV Traub-infected CXCR3- or CXCL10-deficient mice, suggesting that there may be other determinants of disease or redundancy (9,10). While the exact mechanisms that are involved in the protection in these mice are unclear, some data suggest a possible distinguishing role for IFN-γ. While as discussed above, lack of IFN-γ does not alter disease outcome following IC infection of mice with LCMV-Armstrong, this has been reported not to be the case for LCMV-Traub (28). IFN-γ-deficient mice infected IC with LCMV-Traub are protected from lethal disease, indicating that IFN-γ plays a central role in the pathogenesis of LCM with this strain of LCMV. Interestingly, while we observed similar levels of IFN-γ in the CNS of LCMV-Armstrong-infected CXCR3-deficient mice, IFN-γ levels were reduced in CXCR3-deficient mice infected with LCMV-Traub (10).
CXCR3 and/or the ligands CXCL9 or CXCL10 are involved in the trafficking to and/or positioning of activated T cells in the CNS in a number of murine viral infection models. However, in this study we showed that although there is robust expression of the Cxcl9 and Cxcl10 genes and proteins in the CNS following IC infection with LCMV-Armstrong, disruption of the CXCR3/CXCL9-CXCL10 axis by gene-targeted inactivation of CXCR3 did not significantly alter the development of immune pathology, viral load, or survival of the mice.
Conclusion
Our findings indicate that despite similar immunopathogenetic mechanisms in which antiviral CD8+ T cells are crucial, whether or not CXCR3 signaling has a role in LCM appears to depend upon the infecting strain of LCMV.
Footnotes
Acknowledgments
We thank Jane Radford (Department of Pathology, University of Sydney, New South Wales, Australia) for expert technical assistance, and Dr. Bao Lu, Children's Hospital and Harvard Medical School, Boston, for providing CXCR3 KO mice. This work was funded by National Institutes of Health grants NS044905 and NS036979, and a start-up grant from the University of Sydney to I.L.C. M.J.H. and M.M. were postdoctoral fellows from the Deutsche Forschungsgemeinschaft (HO3298/1-1 and Mu17-07/3-1, respectively). M.M. was also supported by the fund “Innovative Medical Research” of the University of Münster Medical School, Münster, Germany. S.L.C. was supported by an Endeavour International Postgraduate Research Scholarship and International Postgraduate Award from the University of Sydney.
Disclosure Statement
No competing financial interests exist.