
Abstract
Prostaglandins (PGs) play an important role in pulmonary physiology and various pathophysiological processes following infection. The initial step in the biosynthesis of PGs is regulated by two distinct cyclooxygenase enzymes, cyclooxygenase-1 (COX-1) and COX-2. The goal of this study was to investigate the pulmonary cellular localization and distribution of COX-1 and COX-2 in a neonatal lamb model following respiratory syncytial virus (RSV) and parainfluenza virus 3 (PI3) infection, organisms that also cause significant respiratory disease in children. No significant differences were seen in pulmonary COX-1 expression at various microanatomical locations following RSV or PI3 infection compared to controls. In contrast, COX-2 was upregulated following RSV and PI3 infection. Strong expression was restricted to bronchial and bronchiolar epithelial cells and macrophages, while minimal expression was present in the same microanatomical locations in the uninfected lungs. Other microanatomical locations in both the controls and the infected lungs lacked expression. This work suggests that during RSV or PI3 infection: (1) COX-1 cellular expression is not altered, (2) COX-2 cellular expression is upregulated in airway bronchiolar and bronchial epithelial cells and macrophages, (3) respiratory epithelium along with macrophages are important microanatomical compartments regulating the host inflammatory response during viral infection, and (4) COX-2 may be a potential target for RSV and PI3 therapy.
Introduction
In the lung, COX pathways are among the many different mediators involved in various physiological and pathophysiological processes, such as regulation of vascular tone (20), pulmonary fibrosis and tissue remodeling (45), surfactant homeostasis (2), alveolar macrophages and their role in pulmonary defense (31,46), bronchial mucus secretion (38), and the pathogenesis of asthma and chronic obstructive pulmonary disease (COPD) (29,33). Additionally, marked increases in prostaglandin E2 (PGE2) synthesis and upregulation of COX-2 have been reported in acid-induced epithelial injury (6), suggesting that COX plays an integral role in the pulmonary system.
Both respiratory syncytial virus (RSV) and parainfluenza viruses (PIs) are pathogens of human infants and perinatal lambs. RSV and PI are leading viral causes of respiratory disease in infants in their first year of life, and cause morbidity and mortality in transplant patients and older adults (15,41). Bovine and ovine strains of both RSV and paramyxoviruses have high homology to human strains and induce identical pulmonary lesions (9,16,17,23,25,27). Several proinflammatory cytokines and chemokines can be detected in patients with RSV and PI infection (5,7,15,21). Increased levels of PGE2 in the plasma or endotracheal aspirates have been demonstrated in animals and infants infected with RSV (14,37). In addition, RSV-induces PGE2 production in human alveolar type II-like epithelial cells (26). However, to the best of our knowledge, COX cellular expression and distribution following RSV or PI3 infection in human or animal models of RSV or PI3 pulmonary infection has not been evaluated.
Therefore, we investigated the pulmonary microanatomical location and cellular expression of COX-1 and COX-2 in lung tissues obtained from a lamb pulmonary model infected with RSV or PI3. This study is the first reported analysis of the pulmonary microanatomical expression of COX-1 and COX-2 in a neonatal lamb model of paramyxoviral infection.
Materials and Methods
Experimental animal model, respiratory viruses, and animal inoculations
A total of 20 neonatal lambs (3–5 d of age) were used in the study. These included: (1) control (saline) inoculated (n = 8), (2) respiratory syncytial virus–infected (bovine strain) lambs (n = 8), and (3) parainfluenza virus-3–infected (ovine strain) lambs (n = 4). The PI3 viral inoculum consisted of infectious supernatant prepared from a culture of ovine fetal turbinate cells previously infected with ovine PI3 virus strain DH-1, and the lambs received 10 mL of 106.9 tissue culture infective doses50 (TCID50) per milliliter intratracheally, and the lambs were inoculated with 10 mL of PI3. The RSV strain 375 was grown on bovine turbinate cells to a concentration of 107 TCID50 per milliliter, and the lambs received 10 mL of RSV (16,27). The control group was given saline.
Pulmonary tissues
On day 6 of infection, the time of peak lesion severity for both RSV and PI3 (16,27), the lambs were euthanized with an intravenous injection of sodium pentobarbital. The thorax was opened and the lungs were then removed from for tissue collection. The lungs were consistently sampled from the left and right cranial, middle, and caudal lobes. The tissues were fixed in 10% neutral buffered formalin for 24 h and embedded in paraffin wax. Sections were cut (3 μm) and stained immunohistochemically with antibodies to COX-1 and COX-2.
COX-1 and COX-2 expression by immunohistochemistry
Immunohistochemical (IHC) staining was conducted as previously described (31). Sections (3-μm thick) were mounted on positively-charged glass slides, dried, and then loaded on the automated immunostainer at 37°C, and we used a Ventana Discovery XT device (Ventana Medical Systems, Tucson, AZ) to analyze COX-1 and COX-2 expression. The slides were deparaffinized and rehydrated. The sections were incubated for 30 min with serum-free DakoCytomation protein blocker (Dako Corporation, Dako, CA), and then rinsed and incubated for 4 min with avidin-biotin blocking solution (Ventana Medical Systems). Heat-induced epitope antigen retrieval was completed using a Ventana specialty solution (pH = 8) (Ventana Medical Systems).
Automation included exposure to 100 μL of primary anti-COX-1 (1:200) or anti-COX-2 (1:50), rabbit polyclonal antibody (Cayman Chemical, Ann Arbor, MI) diluted using reagent diluent (Ventana Medical Systems) at 37°C for 60 min. Then 100 μL of the appropriate anti-rabbit biotinylated IgG (H + L) linking solution (Vector Laboratories, Burlingame, CA) was applied to each section at 1:1000 dilution for 30 min at room temperature. The sections were again rinsed and allowed to react with 100 μL of diaminobenzidine (DAB detection kit) substrate solution (Ventana Medical Systems) for 8 min, followed by counterstaining with hematoxylin then bluing reagent for 4 min each, removed from the autostainer, washed in warm water, dehydrated using graded alcohol, cleared in xylene, and cover-slipped. Control reactions included: (1) sections incubated with the omission of primary antibody and processed as mentioned above, and (2) sections incubated with normal rabbit serum instead of the primary antibody and processed as above. The IHC distribution and intensity scoring system consisted of: (–) = no expression, (+) = earliest weak minimal detectable expression; (++) = mild expression in up to 30% of pulmonary cell types; (+++) = moderate expression in 30–60% of pulmonary cell types; and (++++) = strong expression with >60% of pulmonary cell types.
Results
Clinical signs, gross pathology, and histopathology lesions from these animals were published previously (16). Briefly, both PI3- and RSV-infected animals had increased temperatures compared to controls. Gross pathology lesions were characterized by multifocal areas of red to light-red areas of consolidated regions (often 1–3 mm in size). Microscopically, the lambs had bronchiolitis characterized by slightly dilated bronchiolar lumens partially filled with neutrophils, cell debris, and occasional sloughed epithelial cells, with mild multifocal erosions. Occasional syncytial cells were present in multifocal bronchiolar lumens. Bronchiolar adventitia and alveolar septa contained mild infiltrates of lymphocytes, plasma cells, and occasional macrophages. Alveolar lumens were often partially collapsed and partially filled with seroproteinaceous fluid, alveolar macrophages, and cell debris.
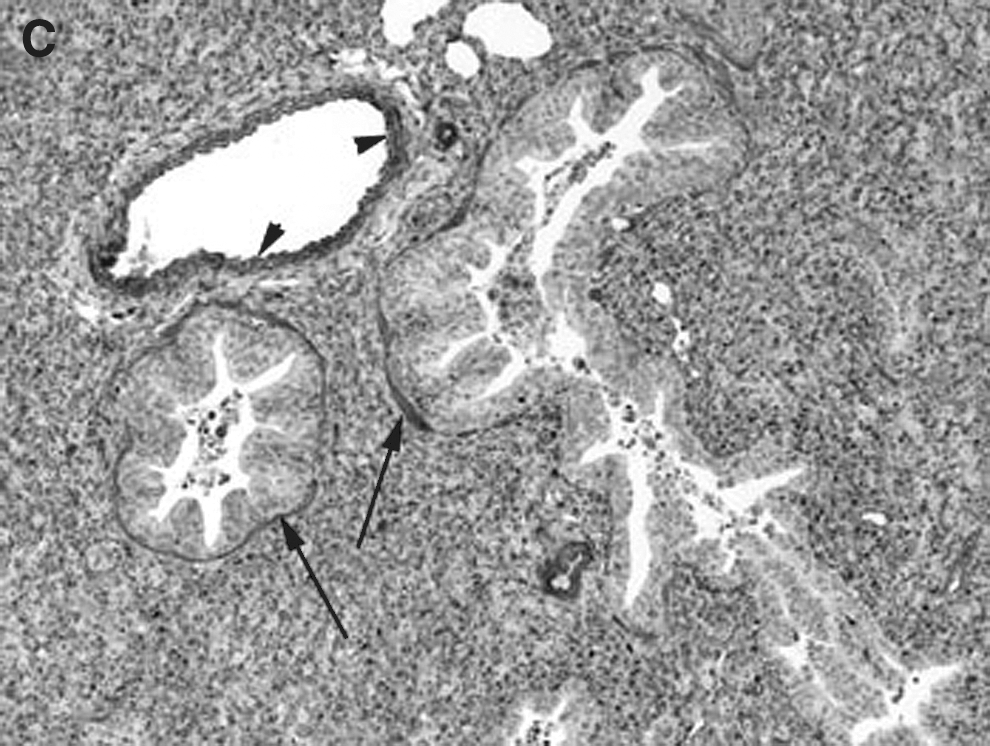
The pulmonary cellular expression and distribution of COX-1 and COX-2 during RSV and PI3 infection are summarized in Tables 1 and 2. Staining intensity ranged from none (–) to strong (++++). Strong (++++) COX-1 expression was present in macrophages, bronchial and bronchiolar smooth muscle cells, vascular endothelial cells, and vascular smooth muscle cells, in lungs from control, RSV-infected, and PI3-infected neonatal lambs (Fig. 1A, B, and C). Intense expression also occurred at sites of inflammation and injury, and correlated with the degree of inflammation and infection. Moderate (+++) COX-1 staining was present in alveolar septa in control and PI3-infected lungs. Alveolar septa from RSV-infected lungs had strong (++++) COX-1 expression. Mild (++) COX-1 expression was present in bronchial and bronchiolar epithelial cells from RSV-infected lungs, while minimal (+) expression was present in lungs from either control or PI3-infected lungs. Bronchiolar epithelium in sections from control and PI3-infected lungs had minimal staining, while mild staining was present in sections from RSV-infected lungs.
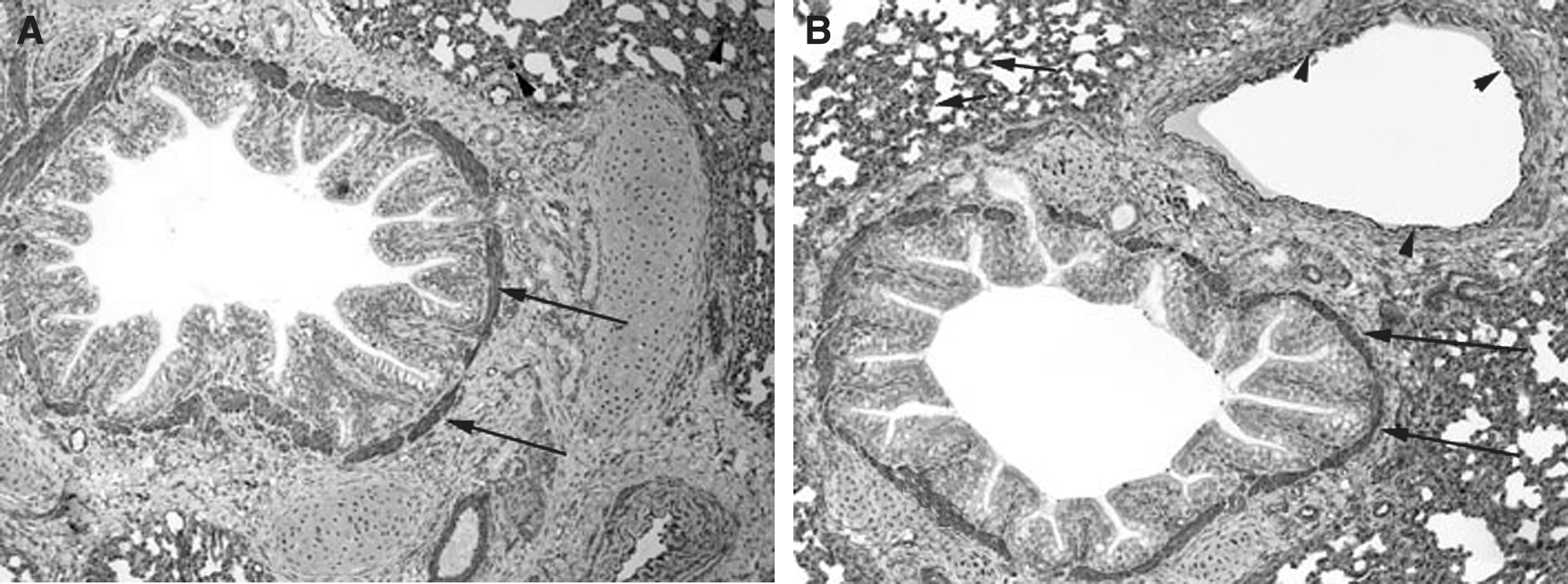
(
(−) = No staining; (+) = minimal expression; (++) = mild expression; (+++) = moderate expression; (++++) = strong expression.
Abbreviations: EC, endothelial cells; SMC, smooth muscle cells.
(−) = No staining; (+) = minimal expression; (++) = mild expression; (+++) = moderate expression; (++++) = strong expression.
Abbreviations: EC, endothelial cells; SMC, smooth muscle cells.
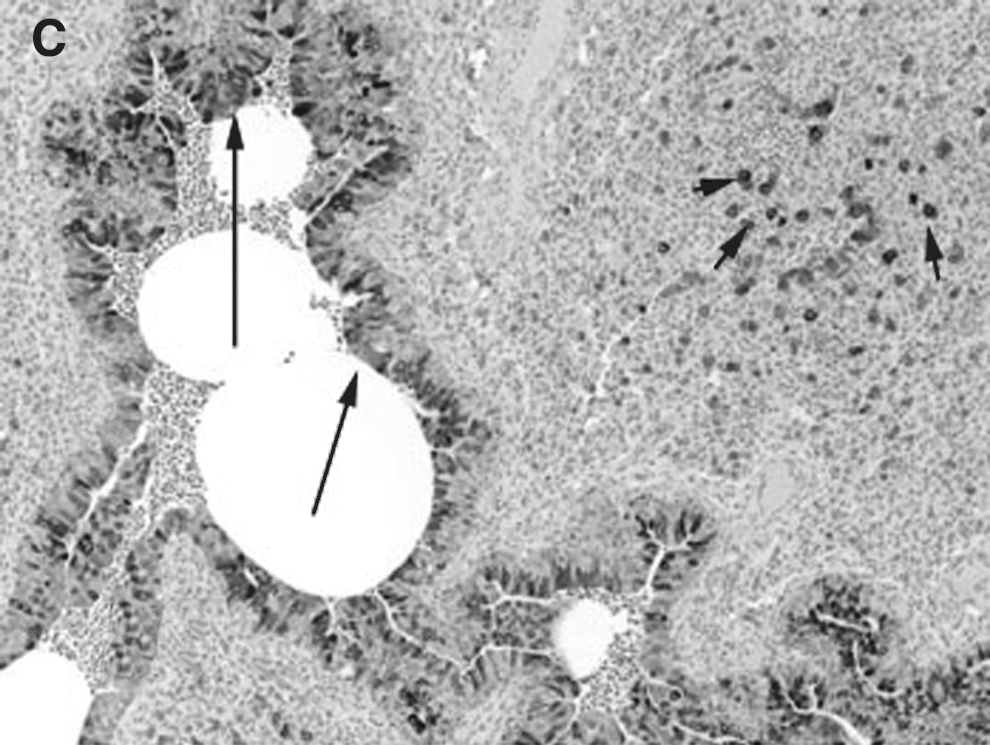
COX-2 expression was minimally present in macrophages from control lungs, while strong (++++) expression was present in macrophages in RSV- and PI3-infected lungs (Fig. 2A, B, and C). Strong (++++) COX-2 expression was present in bronchial epithelium from both PI3-infected and RSV-infected lungs. Bronchial epithelium from control lungs had minimal COX-2 expression. Similarly, strong (++++) COX-2 expression was present in the bronchiolar epithelium of both RSV-infected and PI3-infected lungs, with control lungs had minimal (+) COX-2 expression. Alveolar septa, bronchial and bronchiolar smooth muscle cells, and vascular endothelial and smooth muscle cells lacked COX-2 expression in control and infected lungs (–). Intense expression was present at sites of infection and inflammation and correlated with the degree of inflammation and infection.
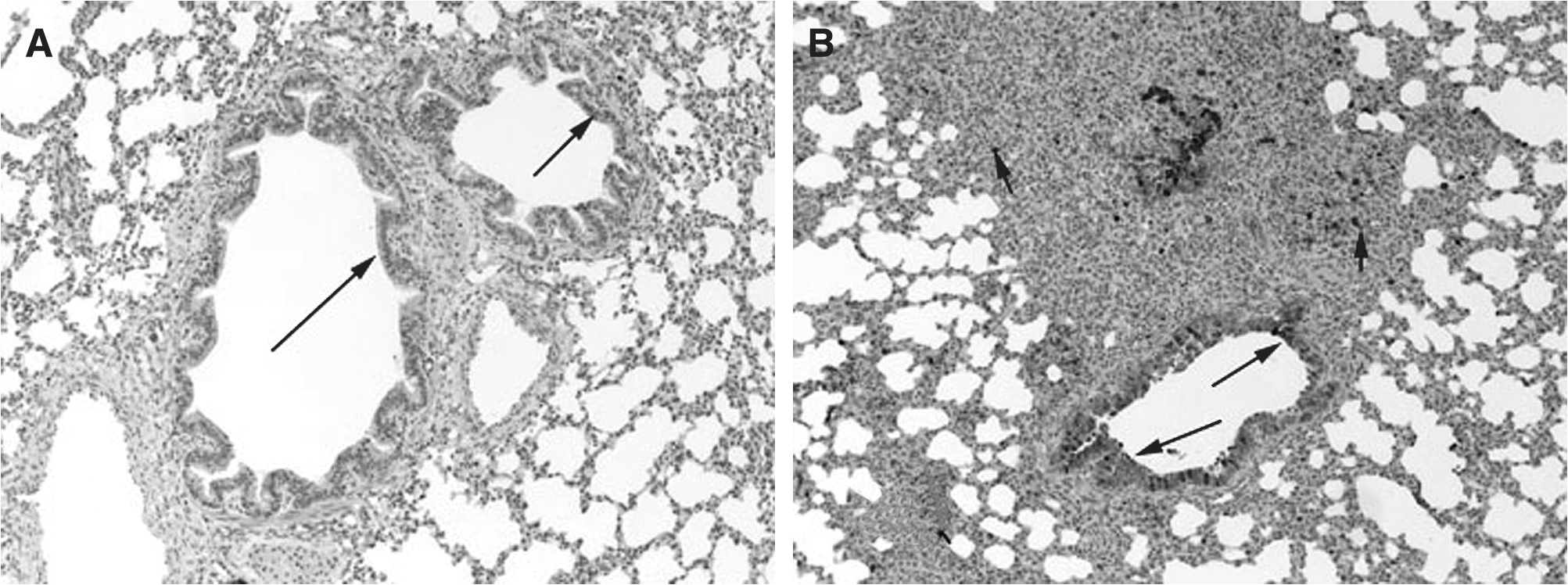
(
Discussion
Pulmonary epithelial cells play an integral role in airway homeostasis, perform numerous biological functions, and represent the first line of defense against infection (39). Prostanoids, generated by COX, are present in high concentrations during various pulmonary disease conditions, such as acute respiratory distress syndrome (ARDS), asthma, COPD, and sepsis (13,29,42). In addition, increased COX-2 expression has been demonstrated in various viral infections (36). Prostanoids are important mediators in both normal and pathological pulmonary functions. All microanatomical locations in control, RSV-infected, and PI3-infected lungs had some degree of COX-1 expression in our study. This finding is consistent with other studies demonstrating COX-1 expression in various pulmonary microanatomical locations in normal human lungs (18). COX-1-dependent prostanoid generation has also been associated with regulation of bronchial tone (11). In lungs from normal animal models, COX-1 was present in bronchiolar epithelium and smooth muscle, alveolar macrophages, endothelial cells, and vascular smooth muscle cells of the rat (11), and in vascular endothelium and alveolar epithelial cells in the non-human primate (24). In lungs of sheep, COX-1 was present in endothelial cells and airway epithelium, and have critical involvement in vasodilatation, bronchodilation, and surfactant synthesis (8). The strong COX-1 expression in macrophages seen in our study is in accordance with results of other studies in rats, indicating that COX-1 was largely responsible for the enzyme activity displayed by alveolar macrophages (46).
Bronchial epithelial cells are known to play an integral role in airway defenses via mucociliary clearance, and they also constitute a mechanical barrier. In addition, bronchial epithelial cells produce and release biologically active compounds, including lipid mediators, growth factors, and a variety of cytokines important in the pathogenesis of airway disorders (40). Some in-vitro studies have demonstrated that cultured human bronchial epithelial cells express COX-2 constitutively (3). In addition, COX-2 expression was found in bronchial epithelium obtained from normal human subjects (10). RSV is a leading cause of respiratory infection and hospitalization in young children, especially those born preterm (17). Likewise, PI1 and PI3 are important causes of seasonal respiratory disease in human infants (17). Sheep are also susceptible to ovine and bovine strains of RSV and ovine strains of PI3. All of these strains have a very high degree of homology and induce similar lung lesions in both perinatal lambs and humans (9,16,17,23,27). The role of prostanoids in modulating RSV or PI3 infection in vivo is unknown. A slightly higher level of COX-1 expression was present in bronchial and bronchiolar epithelial cells from RSV-infected lungs compared to controls and PI3-infected lungs. The marked increase in COX-2 expression in pulmonary bronchial and bronchiolar epithelium and macrophages following RSV or PI3 infection found in our study is consistent with the known role of respiratory epithelia as the first line of defense during infection.
The airway epithelium forms a continuous barrier that limits the access of luminal substances to the systemic circulation (43). In a human airway epithelial cell culture system, COX-2 induction has been observed in the setting of inflammatory cytokine stimulation (44). Significant induction of COX-2 expression and activity was also observed in an experimental mouse model of acute lung injury (12). Minimal COX-2 expression was present in bronchial and bronchiolar epithelial cells from control animals, but was increased in RSV- and PI3-infected animals in our study. This observation corroborates results of other studies that have demonstrated low levels of COX-2 in untreated human airway epithelial cells in culture (28). It has been shown in in-vitro human alveolar type II-like epithelial cells that RSV infection induces a time-dependent increase in COX-2 expression, is a potent inducer of PGE2, and that viral replication is required for PGE2 secretion (26). In addition, strong induction of COX-2 expression during RSV infection in vitro, in human lung alveolar epithelial cells, and in vivo in lungs of cotton rats infected with RSV has been demonstrated (32). However, the exact pulmonary cellular source of the increased COX-2 was not determined in the study by Richardson et al. (32). The lack of COX-2 expression in pulmonary vascular endothelial cells found in our study is consistent with other in-vitro studies in which no COX-2 was detected in these cells under basal conditions (22).
The marked increase in COX-2 expression in macrophages is consistent with the finding that alveolar macrophages are a major source of PGE2 (19). Macrophages are postulated to be the primary cell type expressing COX-2 and producing PGs at sites of inflammation (34). In a mouse model of oxygen-induced ARDS, COX-2 was expressed in alveolar macrophages (1). Increased COX-2 levels in macrophages have been demonstrated in humans after rhinovirus infection (35). COX-2 was induced in RSV-infected cotton rat alveolar and peritoneal macrophages (32). In addition, human cord blood-derived macrophages and dendritic cells have been shown to secrete PGE2 following exposure to RSV (4).
Conclusion
Collectively, the findings in this work suggest that in an in-vivo neonatal lamb model: (1) COX-1 expression is not altered after RSV or PI3 pulmonary infection, (2) COX-2 expression is upregulated in bronchiolar and bronchial epithelial cells and macrophages after RSV and PI3 infection, (3) respiratory epithelia along with macrophages are important microanatomical compartments regulating the host inflammatory response during viral infection, and (4) COX-2 may be a potential target for RSV and PI3 therapy.
Footnotes
Acknowledgments
The work is supported in part by National Institutes of Health grants no. R01 AI062787 (M.R.A.) and KO8AI055499 (D.K.M. and M.R.A.).
Author Disclosure Statement
This work was supported by Pfizer.