
Abstract
The receptor-binding domain (RBD) of severe acute respiratory syndrome coronavirus (SARS-CoV) spike (S) protein plays an important role in viral infection, and is a potential major neutralizing determinant. In this study, three hybridoma cell lines secreting specific monoclonal antibodies against the RBD of the S protein were generated and their exact binding sites were identified. Using yeast surface display, the binding sites of these antibodies were defined to two linear regions on the RBD: S337–360 and S380–399. Using these monoclonal antibodies in phage peptide library screening identified 10 distinct mimotopes 12 amino acids in length. Sequence comparison between native epitopes and these mimotopes further confirmed the binding sites, and revealed key amino acid residues involved in antibody binding. None of these antibodies could neutralize the murine leukemia virus pseudotyped expressing the SARS-CoV spike protein (MLV/SARS-CoV). However, these mAbs could be useful in the diagnosis of SARS-CoV due to their exclusive reactivity with SARS-CoV. Furthermore, this study established a feasible platform for epitope mapping. Yeast surface display combined with phage peptide library screening provides a convenient strategy for the identification of epitope peptides from certain antigenic proteins.
Introduction
The SARS-CoV genome encodes four structural proteins: the spike (S), membrane (M), envelope (E), and nucleocapsid (N) proteins (29,37). The S protein of SARS-CoV, a large class I transmembrane glycoprotein consisting of an S1 domain (residues 15–680) and an S2 domain (residues 681–1255), mediates the attachment of virions to susceptible cells and fusion of the viral and cellular membranes (22,23). Angiotensin-converting enzyme 2 (ACE2) has been identified as a functional receptor for SARS-CoV (27), and a fragment located in the middle region (residues 318–510) of the S1 subunit is the corresponding receptor-binding domain (RBD) (45,46). The S protein of SARS-CoV is not only essential for the process of viral infection, but among all of the structural proteins, is also the major surface antigen and inducer of neutralizing antibodies (2,5,6,20,42,48). Recent studies demonstrated that in addition to the inactivated SARS-CoV vaccine that is capable of eliciting high titers of S-protein-specific antibodies that block receptor binding and virus entry, the RBD alone can induce highly potent neutralizing antibodies (19,21). These data suggest that the RBD of S protein is a major neutralization determinant in the inactivated SARS vaccine.
The RBD of S protein has the potential to be developed as a subunit vaccine, since it induces higher titers of antibodies to block S protein binding with its receptor to neutralize SARS-CoV infection, and has a lower risk than inactivated viruses or live attenuated virus vectors expressing the full-length S protein of SARS-CoV (14,30,33,43). However, caution should still be taken in developing subunit vaccine candidates based on the RBD of S protein. For example, fragment S324–488 within RBD was identified as a functional region responsible for IL-8 production in lung cells, which initiates the inflammatory response and associated lung lesions (7). The ACE2-binding domain mediates antibody-dependent enhancement of civet SARS-CoV-like virus entry (49). A clear-cut analysis of the functional domains of the viral protein responsible for the harmful or beneficial effects on humoral immunity would be a prerequisite for the future use of these vaccines in immunoprophylaxis. Although the RBD is a small part of full-length S protein, only limited knowledge of its structure and function is currently available. Monoclonal antibodies (mAbs) against SARS-CoV, especially RBD-specific mAbs, will be useful tools for probing the antigenic structures and functional regions of RBD in the context of antigen-antibody interactions, and will therefore play a vital role in early diagnosis and pathological studies of SARS.
In this report, we generated three hybridoma cell lines secreting specific mAbs against the RBD of S protein from mice immunized with fragment S249–667 containing RBD. Their exact binding sites were identified by combining yeast surface display with phage peptide library screening strategies, and their neutralizing activity was tested against the infectivity of the murine leukemia virus pseudotyped expressing the SARS-CoV spike protein (MLV/SARS-CoV).
Materials and Methods
Expression, purification, and identification analysis of recombinant S1 fragments
The plasmid used in this procedure encoded the full-length cDNA sequence of the S protein of SARS-CoV strain BJ01 (GenBank accession no. AY278488), and has been described previously (47). Three DNA fragments encoding the amino acid residues 14–248, 249–445, and 249–667 of the SARS-CoV S1 protein were amplified from this plasmid using PCR. These gene fragments were digested with BamH I and Kpn I and ligated into the corresponding restriction sites in pEQ-30 to prepare pEQ-30/S14–248, pEQ-30/S248–445, and pEQ-30/S249–667, respectively. The plasmids were then transformed into competent M15 cells, and the transformed cells were cultured and induced following the protocol described in the Qiagen manual. Recombinant fragments with 6 histidine residues at their carboxyl terminal were purified using Ni resin under denaturing conditions according to the manufacturer's instructions. The recombinant fusion proteins were subsequently detected by Western blot analysis with sera of convalescent-phase SARS patients and healthy donors, providing primary antibodies. Horseradish peroxidase-labeled goat anti-human IgG was used as the secondary antibody, and aminoethyl carbazole signal solution chromogen (Zymed Laboratories, San Francisco, CA) was used for signal detection.
Immunization of animals
Female 6-week-old BALB/c mice were immunized in their rear footpads and subcutaneous tissues with purified recombinant S249–667 (50 μg protein per mouse) emulsified with an equal volume of complete Freund's adjuvant for the first injection, and with incomplete Freund's adjuvant for the following 3 booster injections. The sera sampled from the tail of the mice after the fourth immunization were tested by ELISA to identify the mouse with the strongest response to S249–667. Three days before the fusion experiment, the identified mouse was injected intravenously with 100 μg of S249–667 protein without adjuvant.
Generation and screening of SARS-CoV-specific hybridoma cell lines
The spleen cell suspensions from the hyperimmunized mouse were fused with NS-1 myeloma cells. After hypoxanthine-aminoprotein-thymidine medium selection, the hybridoma culture supernatants were analyzed for antibodies against recombinant protein S249–667 using ELISA. Positive hybridoma clones were selected and further cloned by limiting dilution until the positive rate reached 100%. The reactivity of mAbs with SARS-CoV-infected cell culture filtrates was assayed using ELISA. The classes and subclasses of mAbs were further determined using a Mouse Monoclonal Antibody Isotyping Kit (Roche Applied Science, Indianaopolis, IN) according to the manufacturer's instructions. Indirect immunofluorescence staining was assessed in Vero cells infected with SARS-CoV, MRC-5 cells infected with human CoV 229E, and BS-C-1 cells infected with human CoV OC43. All experiments with the virus were performed in a biosafety level 3 laboratory.
Construction and transformation of yeast eukaryotic expression plasmids
DNA sequences encoding overlapping fragments of the S1 protein were amplified from plasmid pcDNA3.1/S by PCR with specific primers. The PCR products were digested with BamH I and EcoR I and ligated into a pYD1 vector (Invitrogen, Carlsbad, CA), and digested with same restriction enzymes, resulting in pYD1/Sx-y, in which x and y represent the amino acid numbers of the start and stop positions on the S protein. All of the inserted DNA sequences were confirmed by sequencing. All recombinant fragments were fused into the C-terminus of Aga2 for presentation on the yeast cell surface. The C-terminal V5 epitope and the 6 × His tag of fusion protein were used for detecting the expression of target fragments by fluorescent staining.
The yeasts were transformed using lithium acetate. Briefly, the EBY100 glycerol stock was streaked out on a minimal dextrose plate containing leucine and tryptophan, and then incubated at 30°C until colonies appeared 2 d later. The EBY100-competent cells were prepared with 1 M lithium acetate, 50 μg salmon sperm DNA, and 50% polyethylene glycol admixtures, and co-transformed with plasmids pYD1 and pYD1/Sx-y. The transformed mixtures were spread on leucine-containing minimal dextrose plates, followed by incubation at 30°C for 2–4 d until colonies appeared.
Induction of yeast surface-displayed fusion protein
To express the fusion protein on the EBY100 yeast cell surface, the transformants were grown in glucose-containing medium overnight, and then switched to a medium containing galactose for induction of expression. The experimental procedures described below were applied according to the manufacturer's instructions and a previously published protocol (3). Briefly, a single yeast colony was inoculated into 8 mL of yeast nitrogen-based casamino acid medium containing 2% glucose and grown overnight at 30°C with shaking. When the A600 readings reached 5, the cell cultures were centrifuged at 3000–5000 g for 5 min at room temperature. The cell pellets were resuspended in yeast nitrogen-based casamino acid medium containing 2% galactose to an A600 density of 0.5–1, ensuring that the cells continued to grow in a log-phase pattern. The yeast cells were incubated at 20°C with shaking. The cell cultures were assayed over a 48-h period (at 0, 12, 24, 36, and 4 h) to determine the optimum induction time for maximum display. The maximum display of fusion proteins on the EBY100 cell surface was observed 24–36 h after induction. The optimum displayed yeast cultures were then stored at 4°C for further analysis.
Fluorescent labeling of yeast surface-displayed proteins
For the fluorescent labeling of yeast surface-displayed proteins, 1 × 106 yeast cells were microcentrifuged. The cell pellets were resuspended in ice-cold PBS containing 0.1% bovine serum albumin and washed twice. The cell pellets were then resuspended in PBS containing either the primary antibody against the V5 epitope tags or the SARS-CoV S-protein-specific mAbs and incubated at 4°C for 30 min. The cells were then added to PBS containing FITC-labeled anti-mouse IgG at a titer of 1:50, incubated at 4°C for 1 h in the dark, and then washed twice with ice-cold PBS. Finally, the yeast cells were resuspended in 300 μL of PBS and kept at 4°C for flow cytometry analysis. To distinguish the linear epitopes from the conformational epitopes, the yeast cells were heated to 80°C for 30 min in a thermostatically controlled water bath, and then chilled on ice for 20 min before labeling with antibodies. The specific antibody-binding assay was performed with a FACSCalibur flow cytometer (Becton Dickinson Inc., Franklin Lakes, NJ), and the results were analyzed by CellQuest Macintosh software (Becton Dickinson Inc.). Mouse IgG1 and IgG2a (BD Biosciences, San Diego, CA) were used as isotype controls for S1 mAb and S2/S3 mAbs, respectively.
Western blot
The optimum displayed yeast cultures were harvested and disintegrated using acid-washed glass beads (Sigma-Aldrich, St. Louis, MO). The yeast cell lysates were then mixed with SDS sample buffer containing 100 mM DTT, denatured by heating for 5 min at 100°C, and migrated on 10% SDS-PAGE gels. The gels were electroblotted to PVDF membranes (Bio-Rad, Hercules, CA) using 80 mA for 2 h, then blocked overnight at 4°C in TBS containing 2.5% skim milk. The membranes were incubated overnight at 4°C with blocking buffer containing 0.5 μg/mL of mAbs against the S protein or V5 epitope. The membranes were washed in TBS containing 0.1% Tween 20, and incubated in blocking buffer containing horseradish peroxidase-conjugated rabbit anti-mouse IgG at a 1:6000 dilution for 2 h at room temperature. The blots were then washed in TBST, and signals were detected using a chemiluminescence Western-Light Kit (Tropix, Bedford, MA).
Immunohistochemistry (IHC)
Chinese hamsters were anesthetized with isoflurane and euthanized by cervical dislocation on days 1 and 3 following virus administration (28). The lung tissue was fixed with 10% neutral buffered formalin, embedded in paraffin, and processed for routine IHC detection (avidin-biotin peroxidase complex technique). The mAbs S1, S2, and S3 (1:1000) were used as the detecting antibody, and 3,3′-diaminobenzidine (DAB) 12 as the chromogenic substrate.
Construction and screening of the phage peptide library
A phage-displayed peptide library was constructed as previously described (53). Each phage displays a 12-amino-acid length of peptide as a fusion protein with the N-terminus of gIII coat protein. This library was screened using S1, S2, and S3 monoclonal antibodies as selective molecules. A five-round biopanning was carried out as the routine procedure (17) for each monoclonal antibody, and positive phage clones were selected from the fifth-round eluted phages by ELISA using mAbs as the coating antigen. Single-strand DNA from the selected phage was prepared from overnight cultures by extraction and purification using the M-13 Isolation Kit (Omega, Beijing, China). The nucleotide sequence was determined by the dideoxynucleotide chain termination method using the primer with the sequence 5′-CCCTCATAGTTAGCGTAACG-3′. The amino acid sequence of the insert in the isolated clones was deduced from the nucleotide sequence. These amino acid sequences were further analyzed using the ClustalW (1.82) Multiple Sequence Alignments server (
Neutralization assays
To determine whether the infectivity of the murine leukemia virus pseudotype (MLV/SARS-CoV) could be blocked with mAbs, neutralization assays were performed as previously described (41). QT6/ACE2 cells were infected with MLV/SARS-CoV, that had been preincubated with serial dilutions of mAbs S1, S2, and S3. GFP-positive cells were counted 48 h later by fluorescence microscopy. Neutralizing antibody titers were presented as geometric mean titers of assays performed in triplicate.
Results and Discussion
Generation and characterization of SARS-CoV S-protein RBD domain-specific mAbs
At the beginning, we expressed and purified three protein fragments containing amino acid residues 14–248, 249–445, and 249–667 of the S1 protein, followed by detection of its reactivity with the sera of 12 SARS patients by Western blot assays. The results showed that among the three fragments, only fragment S249–667 containing the RBD domain reacted with the sera (data not shown). These results were in accordance with previous observations that the S protein RBDs of coronaviruses MHV and HCoV-229E contained major antigenic determinants that could induce neutralizing antibodies (4,26). The RBD antigen strongly reacted with the neutralizing antibodies against SARS pseudoviruses bearing S proteins of SARS-CoV (Tor2 strain) in the antisera of mice and rabbits immunized with inactivated SARS-CoV (20), and the antisera from patients convalescing from SARS (21). Furthermore, these neutralizing antibodies could be removed by recombinant RBD from antisera, resulting in significant elimination of the neutralizing activity (8,21). Thus further experiments in this study focused on fragment S249–667.
After immunization with the purified recombinant S249–667 protein, hybridomas producing antibodies that were specifically reactive with the SARS-CoV spike protein were successfully established using splenocytes from the immunized mice. Three mAbs were selected on the basis of their reactivity with the S249–667 protein and SARS-CoV-infected cell culture filtrates by ELISA. The isotypes of the mAbs S1, S2, and S3 were IgG1, IgG2a, and IgG2a, respectively. The antibodies were determined as highly reactive with SARS-CoV-infected Vero E6 cells by indirect immunofluorescence assay, but did not recognize human coronaviruses OC43 and 229E (Table 1), indicating that these mAbs were specific to SARS-CoV, without cross-reactivity with other coronaviruses, and therefore had the potential to be used for diagnosis or treatment of SARS-CoV infection.
SARS-CoV-infected cell culture filtrates were used as the coating antigen for ELISA. The absorbance was measured at 450 nm: +, A450 = 0.2 ∼ 1; ++, A450 = 1 ∼ 2. Indirect immunofluorescence staining was assessed in Vero cells infected with SARS-CoV, MRC-5 cells infected with human CoV 229E, and BS-C-1 cells infected with human CoV OC43.
Abbreviations: P, positive signal; N, negative signal.
Yeast cell surface display of overlapping peptides spanning S249–667
The S protein of SARS-CoV is heavily glycosylated and contains many disulfide bonds (37). In general, it is difficult to express and purify the individual protein domains for large scale structural and functional research via a eukaryotic expression system. However, yeast surface display is a useful method for identifying stable folded protein domains from multidomain extracellular receptors and characterizing the antibody binding epitope, without the need for soluble protein expression and purification (9). Yeast expresses folded, functional proteins due to its protein folding and quality control machinery in the endoplasmic reticulum, which is in contrast to bacterial expression systems (13). Therefore this method was chosen for this study to identify the immunogenic peptides spanning S249–667.
DNA sequences encoding a series of overlapping peptides spanning S252–667 were inserted into the yeast display plasmids for cell surface expression. These fragments were well expressed on the yeast cell surface, as indicated by the reactivity of the C-terminal V5 epitope tag with the anti-V5 antibody (Fig. 1 and Table 2). In this yeast display system, a percentage of cells did not express the protein on their surface, and as a result two histogram peaks occurred. The flow cytometry histogram of the positive yeast display showed a peak with background fluorescence equivalent in intensity to that obtained with negative controls, which also provided a good internal negative control of the labeling (3). The well-expressed yeast displayed fusion proteins, and well-characterized S-protein-specific mAbs were used for antibody epitope mapping in the next experiments.

Yeast cell surface display of overlapping peptides spanning S252–667. Flow cytometry histograms depict the mean fluorescence signal of the antibody-labeled C-terminal V5 epitope tag of the yeast-displayed S protein fragments. The presence of the V5 epitope tag confirms the expression of the full-length polypeptide on the yeast cell surface. Untransformed EBY100 yeast cells were used as the negative control. With the yeast display system, a percentage of cells do not express protein on their surface, resulting in two histogram peaks, which serves as a good internal negative control for labeling. Data are representative of three experiments.
The fluorescence intensities of the various yeast-displayed fusion proteins, confirmed to bind to the 3 mAbs by flow cytometry, were calculated by dividing the fluorescence intensity obtained for the individual fusion proteins by the fluorescence intensity obtained for the yeast-expressing empty vector pYD1. Ratio values ≥2 were considered positive, and a result was considered negative when the ratio <2; a weak but detectable positive signal (+) was given when the ratio was 2–5, an intermediate positive signal (++) was characterized as a ratio of 5–10, and a strong positive signal was present when the ratio was >10.
Antibody epitope mapping of the RBD of SARS-CoV S protein
The three mAbs were used in the same concentration (10 μg/mL) and diluted in PBS to a final volume of 100 μL for the binding assay. All assays were performed in triplicate or more, and the individual fluorescence intensities were obtained from the mean value of the specific yeast-displayed protein. The ratio of the recombinant antigen expression on the yeast surface (RAYS) was calculated by dividing the fluorescence intensity obtained for the individual RAYS by the fluorescence intensity obtained from yeast expressing pYD1. A ratio ≥2 was considered positive (31). Results showed that all three mAbs could bind to fragment S318–520 (Table 2 and Fig. 2), and the antibody-binding sites of all three mAbs are therefore located in the ACE2 receptor-binding domain of the S protein. The antigenic determinant sites on the RBD were identified according to the specific binding of a mAb to the particular recombinant overlapping peptides covering residues 318–520. The profile of mAbs binding to S318–420, S318–408, S318–399, and S380–420 indicates that mAb S1 recognizes a region of residues 318–380, whereas mAbs S2 and S3 recognize fragment S380–399 in the middle of the RBD (Table 2 and Fig. 2). The S1 binding site was further defined as a smaller region of S337–360 according to the binding of fragments S252–339, S252–360, S337–460, and S355–460 (Table 2 and Fig. 2).
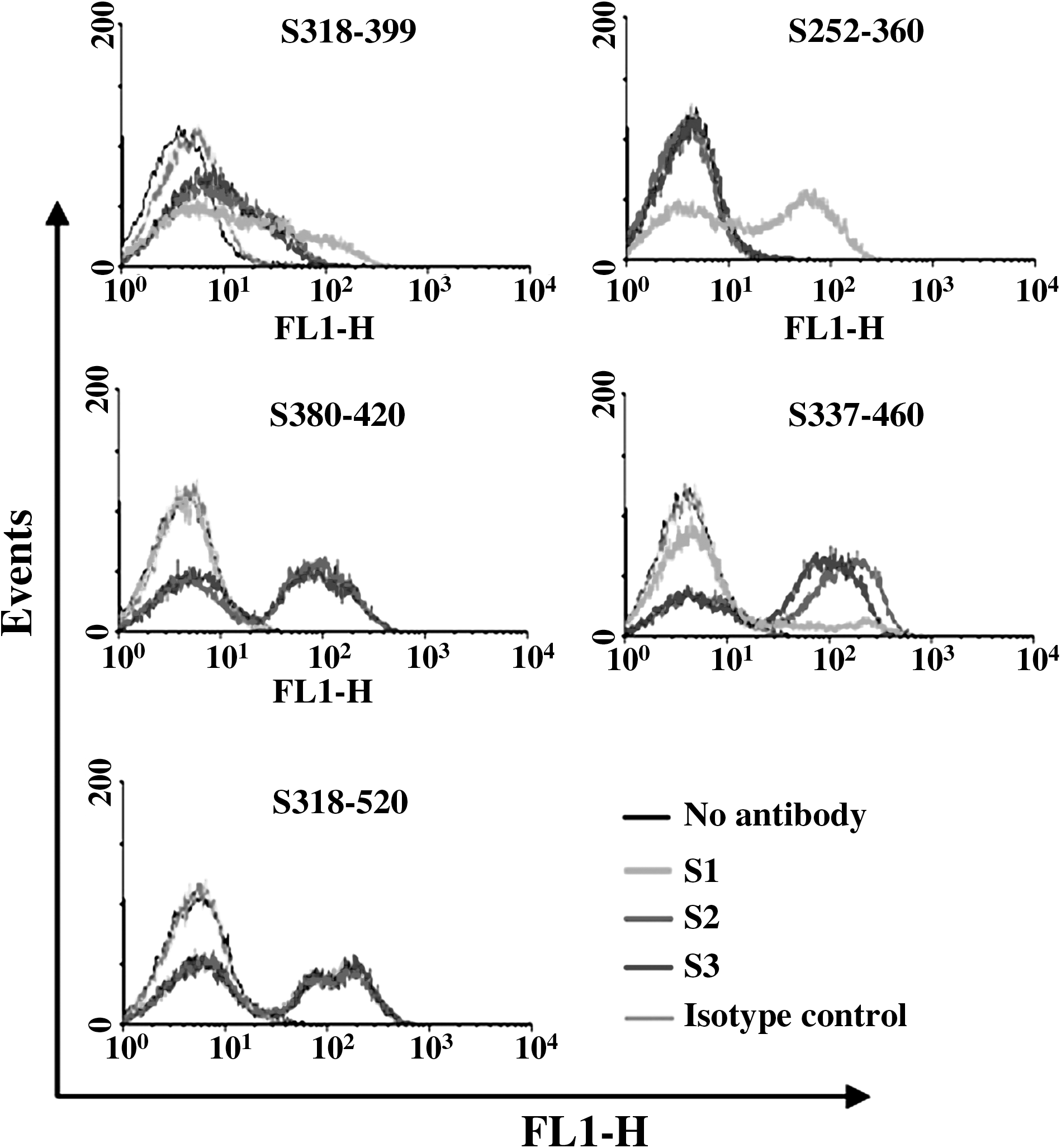
Binding of mAbs S1, S2, and S3 with overlapping peptides spanning S252–667 displayed on the yeast cell surface. Representative flow cytometry histograms depict the mean fluorescence signal of antibody labeling of yeast-displaying fragments. Mouse IgG1 and IgG2a were used as the isotype controls for mAb S1 and S2/S2, respectively.
Differentiation of linear and conformational epitopes
Antibodies developed in this study were categorized as directed against conformational or linear epitopes using the following heat denaturation of the S protein fragment tethered to the yeast cell surface. Yeast cells displaying S318–520 fragments were heated at 80°C for 30 min, chilled on ice, and then assayed for antibody binding by flow cytometry. The detection of the C-terminal V5 epitope tag with mAb anti-V5 confirmed the presence of this fragment on the yeast cell surface, indicating that the proteins and cells were not compromised during the heat treatment (Fig. 3). The three mAbs all retained their binding reactivity upon protein denaturation, demonstrating that these antibodies are specific for linear or continuous epitopes. Results from Western blot analysis also confirmed this conclusion (Fig. 4A). All of these mAbs recognize the S318–520 fusion peptide under reduced and denatured conditions. The anti-S mAbs produced a single band with the S318–520 fusion peptide at exactly the same location (about 40 kDa in relative molecular mass) as the reaction band of anti-V5 antibody.
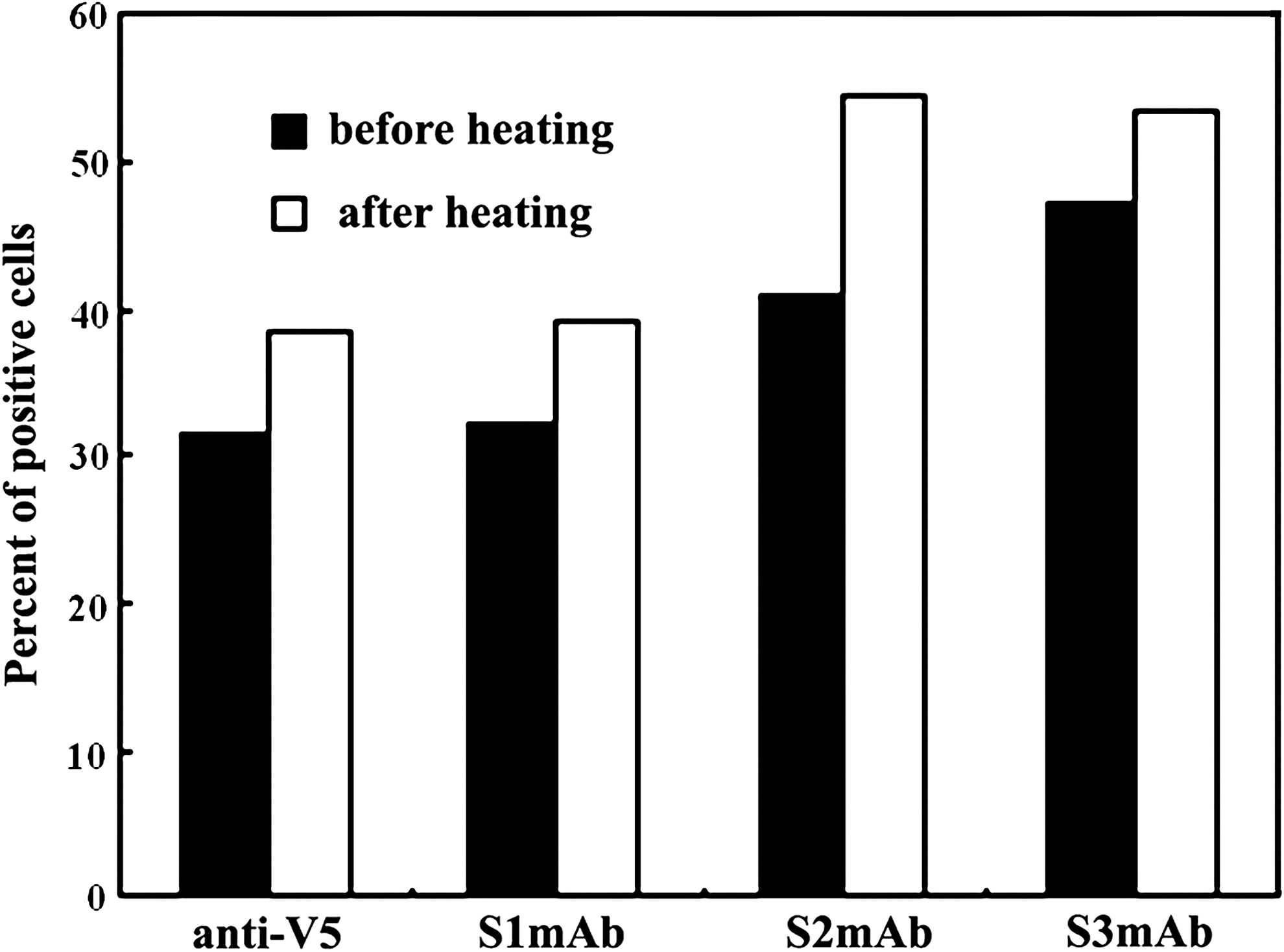
S-protein-specific mAbs recognize linear or continuous epitopes. Antibody labeling was assayed before and after protein denaturation by heating yeast cells to 80°C for 30 min. Anti-V5 mAb labeling (specific for the linear C-terminal V5 epitope tag) demonstrates that heat treatment does not compromise the protein and yeast cell.
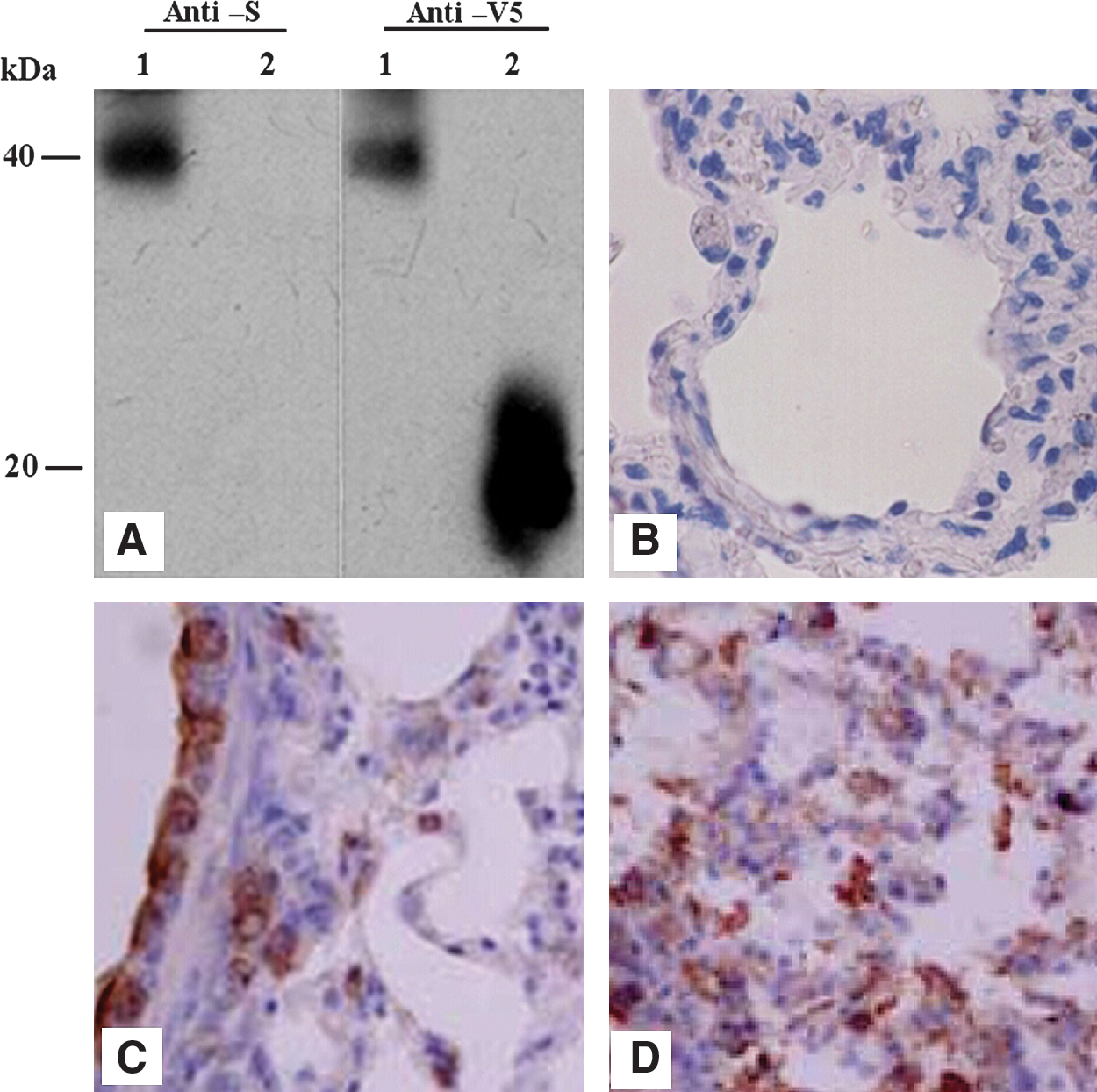
mAbs recognize the intact S protein on the SARS-CoV. (
Considering that the three mAbs can recognize the linear epitopes within S318–520, and that the S318–520 fragment on yeast surface should have a conformational structure, it can be deduced that the epitopes recognized by these mAbs probably reside on the exterior of the conformational structure of native S protein. To this end, we conducted the IHC assays for the three mAbs on the lung tissue sections from SARS-CoV-infected Chinese hamsters (28). The results demonstrated that all the mAbs could recognize SARS-CoVs in the lung tissue on days 1 and 3 following virus administration (Fig. 4B–D), indicating the potential application of these mAbs for the diagnosis or treatment of SARS-CoV infection.
Identification of key amino acid residues at the binding sites using phage display
The phage display random peptide library offers a convenient way to select peptides that mimic the original characteristics of antigen binding to specific antibodies. In the present study, a phage display peptide library composed of 2.1 × 108 independent recombinants was constructed and screened by biopanning using mAbs obtained in this study as selective molecules. After five biopanning procedures, 10 distinct peptides 12 amino acids in length were recovered among the selected positive clones (Fig. 5). The amino acid sequences of these mimotopes and the S protein were analyzed using Clustal W (1.82). The multiple sequence alignments results revealed that five peptides selected using mAb S1 show homology with S330–350, while peptides from phage clones selected using mAb S2/S3 displayed homology with S380–399 (Fig. 5). These data not only further confirm the location of the binding sites obtained using the yeast surface display system, but also provide more detailed information on key binding residues. Thus 340W and 341E are essential for mAb S1 binding, while 390K, 392D, and 395R are important for mAbs S2/S3 binding. These results also indicate that these key binding residues are surface-exposed amino acid residues in the S protein of SARS-CoV.

Amino acid sequences of peptides from the positive phage clones selected using mAbs. Five peptides selected with mAb S1 show homology with S330–350, whereas peptides from phage clones selected using mAb S2/S3 displayed homology with S380–399 (asterisks indicate the preserved binding residues).
Neutralizing effects of mAbs on the infectivity of MLV/SARS-CoV pseudotype virus
SARS-CoV is highly contagious and must be handled under stringent protection, so we utilized the MLV/SARS-CoV pseudotype virus to evaluate the three mAbs. After preincubation with mAbs, MLV (SARS-CoV) showed the same infectivity of QT6/ACE2 cells, as there was no significant reduction in the percentage of GFP-positive cells compared with untreated MLV (SARS-CoV) (data not shown). This result shows that all three mAbs have no neutralizing effects on the infectivity of the pseudotype virus, indicating that either these binding sites are not actively involved in spike-ACE2 binding, or these antibodies cannot compete with ACE2 for binding to the S protein.
Although the RBD of the SARS-CoV S protein is a small, 193-aa fragment, it contains seven cysteine residues, five of which are essential for protein expression and ACE2 association (45). The disulfide bonds between these cysteines may form complex tertiary structures to constitute the multiple antigenic conformations. Recent studies have demonstrated that recombinant RBD consists of multiple conformational neutralizing epitopes that induce highly potent neutralizing antibodies against SARS-CoV (8,18 –21,44). The majority of the neutralizing mAbs against SARS-CoV recognize conformational epitopes, and mAbs recognizing linear short peptides show no neutralizing effects on infectivity of SARS-CoV (1,8,18,39,44,50). Recombinant spike protein vaccines produced by Escherichia coli were not suitable as SARS-CoV vaccine candidates, as they did not generate sufficient protective immunity compared to those generated from eukaryotic systems such as the transfection of cell lines (2,5,6,10,15,24,40,48,51,52,54). Our results are consistent with the above observations. All three mAbs produced in this study recognize linear binding sites and have no neutralizing activity, which may be partially due to the fact that these antibodies were generated using S protein fragments expressed in E. coli. These data suggest that the maintenance of the naïve conformation is necessary for developing vaccines based on the RBD of SARS-CoV S protein. Further characterization is needed to define the neutralization determinants of the RBD of SARS-CoV S protein, and this may provide critical information for developing anti-SARS therapeutics and vaccines. The overlapping fragments displayed on the yeast cell surface in this study are useful agents for mapping conformation-sensitive mAb epitopes.
Conclusion
In summary, we developed three strain monoclonal antibodies against the RBD of the SARS-CoV spike protein, and their binding sites were located at S380–399 and S337–360, as determined using a yeast surface display system and phage display peptide library. Although these mAbs showed no neutralizing activity, they were proven quite to be useful for the diagnosis for SARS-CoV, due to their exclusive reactivity with SARS-CoV over other coronaviruses, and the specific recognition of SARS-CoV in the lung tissue of SARS-CoV-infected hamsters. Furthermore, a platform for epitope mapping was constructed by combining yeast surface display and phage peptide library screening, which is a convenient strategy for the identification of an epitope peptide from a certain antigenic protein.
Footnotes
Acknowledgments
We are grateful to Xiaolan Fu for FACS analysis. This work was supported by the National Key Basic Research Program of China (grants 2001CB510001 and 2003CB514108), the Key Project of the National Natural Science Foundation of China (grant 30490241), and the National Outstanding Young Scientist Foundation of China (grant 30325020).
Author Disclosure Statement
No conflicting financial interests exist.