
Abstract
In addition to genetic factors, environmental triggers, including viruses and other pathogens, are thought to play a major role in the development of autoimmune disease. Recent findings have shown that viral-induced autoimmunity is likely to be genetically determined. In large-scale genetic analyses, an association of interferon induced with helicase C domain 1 (IFIH1) gene variants encoding a viral RNA-sensing helicase with susceptibility to several autoimmune diseases was found. To date, the precise role of IFIH1 in pathogenic mechanisms of viral-induced autoimmunity has yet to be fully elucidated. However, recent reports suggest that IFIH1 may play a role in the etiology of type 1 diabetes. Rare IFIH1 alleles have been shown to be protective against diabetes, and their carriage correlates with lower production of this helicase and its functional disruption. In contrast, upregulation of IFIH1 expression by viruses is associated with more severe disease, and could exacerbate the autoimmune process in susceptible individuals.
Introduction
Despite a growing body of evidence describing associations between viruses and autoimmune disease in genetically prone individuals, clear identification of causative infectious agents has thus far been unsuccessful. A likely explanation is that the link between infections and autoimmunity is more complex than we initially assumed.
However, recently, using genome-wide association studies and other advanced technologies for high-throughput DNA analyses revealed that molecular variants of the IFIH1-GCA-KCNH7 locus on chromosome 2q24.3 could confer predisposition to several autoimmune diseases, such as T1D (95), MS (52), rheumatoid arthritis (RA) (53), and Graves' disease (GD) (97). This genomic region contains the IFIH1 gene, which encodes interferon induced with helicase C domain 1, a viral RNA-activated apoptosis protein with a putative role in sensing and triggering clearance responses in virally-infected cells (113). Therefore, these findings shed new light on our knowledge of virus-induced autoimmunity, suggesting the presence of a shared genetic background in viral autoimmunity. In this review, we consider a putative role of IFIH1 in virus-induced autoimmunity.
Interferon Induced with Helicase C Domain 1 and Viral Autoimmunity
Intracellular mechanisms of viral RNA recognition
Today, double-stranded (ds) RNA is known to represent a molecular intermediary during the replication of many viruses within infected cells, and therefore it is not surprising that the host has evolved several means to detect its presence. The first pathway depends on protein kinase R (PKR). This interferon (IFN)-inducible kinase becomes activated following binding to cytoplasmic dsRNA that results in phosphorylation of the eukaryotic translation initiation factor-α (56). Through this mechanism, PKR is able to inhibit protein translation (and virus replication) within infected cells (110).
A second protein that is stimulated by dsRNA is 2′-5′-oligoadenylate synthetase, an enzyme that activates the endoribonuclease RNase L through the synthesis of short oligoadenylates. Upon activation, RNase L promotes cleavage of both cellular and viral RNAs (22). However, although both pathways become activated by dsRNA and are implicated in antiviral immunity, PKR and RNase L are mainly IFN effectors and are not required for IFN production. Therefore other cellular systems are needed to explain the connection between dsRNA sensing and upregulation of type I IFN.
Such systems include sensory mechanisms mediated by toll-like receptors (TLRs), and DExD/H box-containing RNA helicases. TLRs, numbered 1–11, are receptors that recognize via their leucine-rich repeats derived from microorganisms. TLRs are normally presented at the plasma membrane to detect extracellular pathogen-associated molecular partners. However, a few TLRs, including TLR3, TLR7, TLR8, and TLR9, recognize their ligands in intracellular compartments such as endosomes. Interestingly, the latter TLRs share the ability of nucleic acid recognition of dsRNA (TLR3), single-stranded (ss) RNA (TLR7 in mice and TLR8 in humans), and nonmethylated CpG DNA motifs (TLR9) (2).
The recent characterization of DExD/H box-containing RNA helicases as cytoplasmic viral RNA receptors has defined a novel antiviral pathway, and has also permitted a clearer comprehension of the signals emanating from TLR-dependent and TLR-independent antiviral mechanisms (103).
MDA5 and RIG-I RNA helicases: Distinct roles in sensing viral RNA
IFIH1, which is better known as melanoma differentiation-associated gene 5 (MDA5), belongs to the DExD/H box-containing RNA helicase family, which also comprises two other members: retinoic acid-inducible gene 1 (RIG-I), and LGP2. A full-length IFIH1 cDNA was first isolated from a human placental cDNA library (38). This cDNA, 3365 bp in length, encoded a 1025-amino acid (a.a.) product with a predicted molecular mass of 116.7 kDa (Fig. 1). The protein has two highly conserved structural domains: an N-terminal caspase recruitment domain (CARD), and a C-terminal DExH/D RNA helicase domain closely related to that of RIG-I (38). The CARD motifs of RIG-I and MDA5 activate downstream signaling cascades that result in activation of interferon regulatory factor (IRF)-3, IRF-7, and NF-κB. Recently, site-directed mutagenesis experiments demonstrated that the type I interferon production mediated by full-length MDA5 and RIG-I is independent of the helicase domain catalytic activity (6). Interestingly, the third member of the RNA helicase family, LGP2, contains the helicase domain, but lacks the CARD domain, and therefore is unlikely to activate downstream signaling pathways on its own (113). LGP2, which has been shown to be able to recognize the termini of dsRNA, functions as a negative regulator by interfering with the recognition of viral RNA by RIG-I and MDA5 (46).

(
In antiviral signaling cascades, RIG-I and MDA5 both utilize a common target, an adaptor protein, IFN-β promoter stimulator-1 (IPS-1) (41). IPS-1-deficient mice are defective at producing type I IFNs and proinflammatory cytokines in response to all RNA viruses recognized by either RIG-I or MDA5 (43). These findings therefore indicate that IPS-1 plays an essential role in RIG-I/MDA5 signaling. IPS-1 was also found to associate with TNF-receptor-associated factor (TRAF) 3, that finally results in IRF3- and IRF7-mediated expression of type I IFNs, and NF-κB-directed production of proinflammatory cytokines (82).
The expression of RIG-I, MDA5, and LGP2 is induced at the transcriptional level by retinoic acid, type I IFNs, and dsRNA. Initially these RNA helicases were thought to recognize cytoplasmic dsRNA during viral infection (113). However, the generation of knockout mice lacking either RIG-I or MDA5 revealed that the two helicases are not redundant in their ability to recognize viral RNA. Each helicase was individually dispensable for signaling triggered by West Nile virus (28) and Dengue virus (39), both belonging to the Flaviviridae family, and likely also by rotaviruses and reoviruses (92). RIG-I was essential for the production of IFNs in response to a wide variety of RNA viruses, including paramyxoviruses (e.g., mumps virus, measles virus, parainfluenza viruses, and human respiratory syncytial virus), influenza A and B viruses, vesicular stomatitis virus, human hepatitis C virus, and Japanese encephalitis virus (39,49,79,83,96), whereas MDA5 was critical for specific detection of picornaviruses such as rhinoviruses, echoviruses, encephalomyocarditis virus, Theiler's virus (30), and poliovirus, a prototypical picornavirus (7). However, paramyxovirus protein V shares a region of homology with MDA5, but not RIG-I (70), which is essential for preventing RNA binding, dimerization, and activation of MDA5 (13). This finding could suggest that MDA-5 has been evolutionarily involved with triggering the innate immune response to paramyxovirus infections. On the other hand, the phenomenon of RIG-I cleavage in cells infected with picornaviruses (8,69) provides evidence that RIG-I has also been implicated in the immune response to picornaviruses during evolution.
Thus experimental data suggest that RIG-I and MDA5 utilize similar intracellular signaling mechanisms to induce an anti-RNA virus response. However, the sensory function of these helicases is distinct, since they are induced by different sets of viruses. The difference is likely to arise from the ability of MDA5 to recognize dsRNA that, for example, is generated by cells infected with encephalomyocarditis virus, but not influenza virus (76). On the other hand, RIG-I, but not MDA5, recognizes 5′-triphosphate ssRNA (36,76,83). In recent studies researchers reported an ability of RIG-I to recognize dsRNA, which requires base-paired structures in conjunction with a free 5′-triphosphate to be recognized by RIG-I (60,87). In addition, experiments with polyinosinic:polycytidylic (poly I:C) RNA and the reovirus genome showed that the short segments of dsRNA preferentially activate RIG-I(40), while the longer segments of dsRNA are better recognized by MDA5 (100). However, the mechanism underlying this discrimination between long and short dsRNA of MDA-5 and RIG-I remains to be elucidated.
RNAs from some viruses, such as influenza virus and vesicular stomatitis virus, are 5′-triphosphorylated and uncapped, whereas the 5′ ends of host mRNAs are capped. Thus RIG-I discriminates virus and host RNAs based on the differences in the 5′ ends of their RNAs. The 5′ ends of the RNA genomes and RNA replication intermediates of picornaviruses are not phosphorylated, but are covalently linked to a small protein, VPg (72). Such RNA molecules, therefore, would not be recognized by RIG-I.
Signaling cascade mediated by RIG-I-like helicases to induce an antiviral response
Signal transduction pathways mediated by RIG-I-like helicases have been examined in detail in several recent reviews (9,61,114). Briefly, MDA5 and RIG-I initiate signaling cascades via the interaction between their CARDs and an N-terminal CARD-like structure of IPS-1 (Fig. 2) (41). Interestingly, the mitochondrial localization of IPS-1 is absolutely necessary for downstream signaling (91). The proline-rich region of IPS-1 is able to bind TRAF family members TRAF2, TRAF3, and TRAF6 (88). TRAF3 is an E3 ubiquitin ligase that assembles lysine 63-linked polyubiquitin chains (82). In cells deficient in TRAF3, production of type I IFNs in response to viral infection is severely impaired (64). A deubiquitinase named DUBA (deubiquitinating enzyme A) was shown to suppress RIG-I-dependent signaling through TRAF3 deubiquitination (42). TRAF3 recruits two signaling protein kinases: TANK-binding kinase 1 (TBK1), and inducible IkB kinase (IKK-i, also known as IKK-ɛ) (26). IKK-ɛ is also activated by IFN-β to directly phosphorylate STAT1, thereby controlling a set of IFN-inducible genes, including the dsRNA-activated adenosine deaminase gene (Adar1) (101).
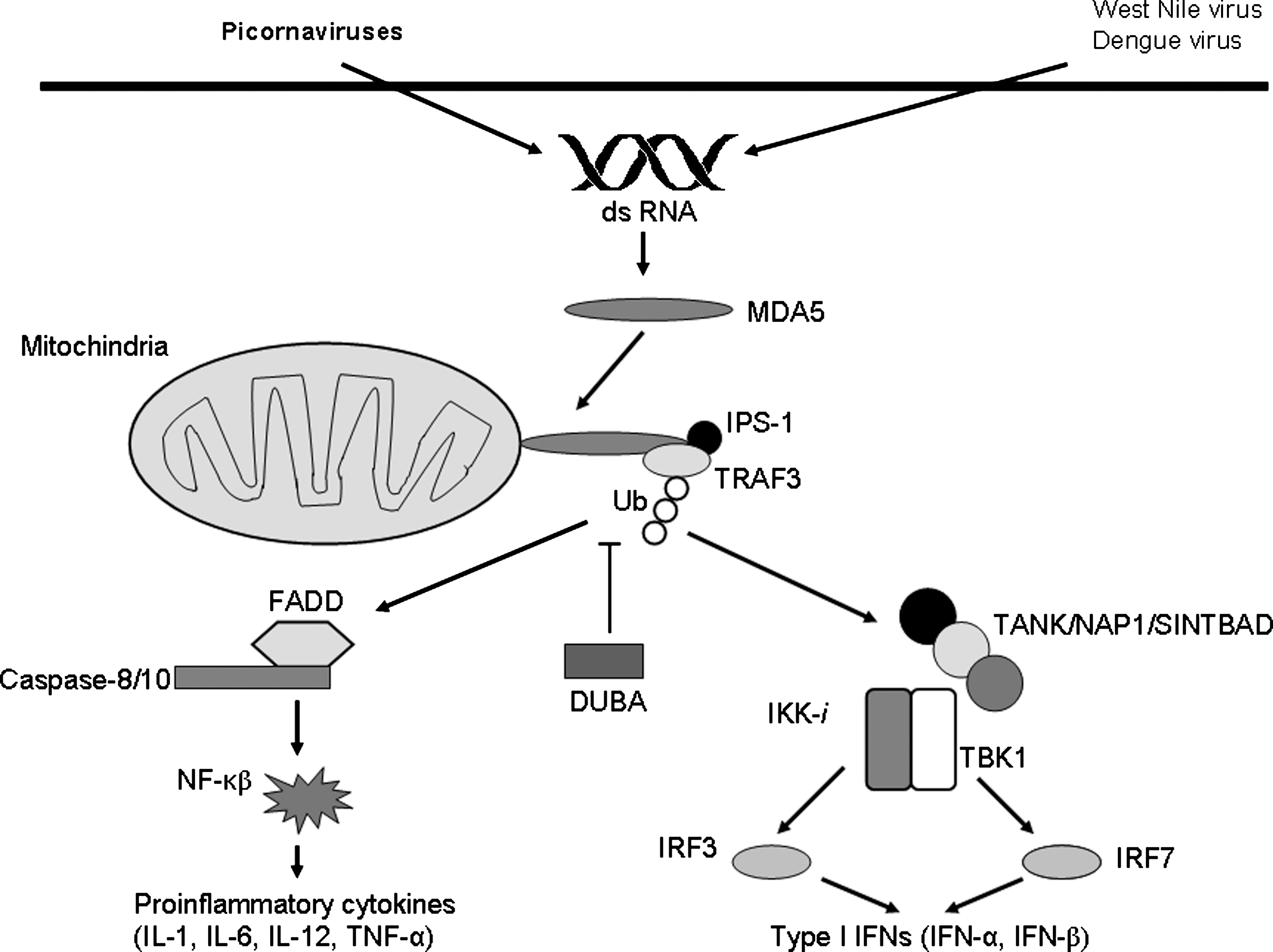
MDA5-mediated recognition of RNA viruses followed by induction of the innate immune response. In the cytoplasm, MDA5 recognizes viral dsRNA. MDA5 specifically senses picornaviruses and shares recognition of West Nile virus and Dengue virus with RIG-I, an RNA helicase that is closely related to MDA5. Viral RNA recognition is accomplished by IPS-1 binding to MDA5. TRAF3 recruitment is required for activation of IPS-1-dependent signaling and K63-type polyubiquitination that is downregulated by DUBA. Subsequently, TRAF3 recruits TANK/NAP1/SINTBAD and TBK1/IKK-i, which phosphorylates IRF-3 and IRF-7. IRFs then translocate to the nucleus to induce expression of type I IFNs. IPS-1 also activates NF-κB through a FADD- and caspase-8/10-mediated pathway, that results in the production of proinflammatory cytokines (DUBA, deubiquitinating enzyme A; FADD, Fas-associated via death domain; IFN, interferon; IKK-i, inducible IkB kinase; IL, interleukin; IPS-1, interferon-β promoter stimulator 1; IRF, interferon regulatory factor; MDA5, melanoma differentiation-associated 5; NAP1, nucleosome-assembly associated protein 1; NF-κB, nuclear factor-κB; TBK1, TANK-binding kinase 1; TNF-α, tumor necrosis factor-α; TRAF3, TNF receptor-associated factor 3; SINTBAD, similar to NAP1 TBK1 adaptor; Ub, K63-polyubiquitination).
Both kinases form a complex with TRAF family member-associated NF-κB activator (TANK), nucleosome assembly-associated protein 1 (NAP1), and similar to NAP1 TBK1 adaptor (SINTBAD) (44–46). Phosphorylation of IRF-3 and IRF-7 by TBK1 and IKK-ɛ induces the formation of homodimers and/or heterodimers (35), which translocate into the nucleus and bind to IFN-stimulated response elements, resulting in the expression of type I IFN genes and a set of IFN-inducible genes. Additionally, FAS-associated death domain-containing protein (FADD) interacts with caspase-8, caspase-10, and IPS-1, and the FADD-dependent pathway is responsible for the activation of NF-κB downstream of IPS-1 (Fig. 2). Both caspases were cleaved during dsRNA stimulation, and overexpression of a cleaved form of these caspases activated NF-κB. Knockdown of caspase-10 or caspase-8 in a human cell line resulted in a reduction in inflammatory cytokine production (99).
IFIH1 and type 1 diabetes mellitus
An association between the IFIH1 gene and T1D was first reported by Smyth et al. (95), who performed genome-wide association studies in affected European families, and a large population cohort of U.K. Caucasians, totaling over 10,000 subjects. Several single nucleotide polymorphisms (SNPs) located within the IFIH1 gene region showed an association with diabetes, with marker rs1990760 (NP_071451.2:pA946T) being the most strongly associated. This polymorphism is an adenine-to-guanine substitution located at exon 14, and resulted in an amino acid change of alanine to threonine at codon 946. The minor Thr-encoding allele was associated with a lower risk of T1D (odds ratio [OR] = 0.86; Poverall = 1.42 × 10−10).
With the exception of two studies (2,42), an association between IFIH1 and T1D in Caucasians was then replicated in several other studies (16,47,62,78). The marker rs1990760 showed an association with T1D in U.S. Caucasians from Georgia, along with three other SNPs: rs374517A/G (NP_071451:pH843R), rs2111485A/G, and rs13422767 (NT_005403.16:g13309677G>A) (47). Of those, only rs374517 resides in the coding region of IFIH1, changing histidine to arginine at position 843 in exon 13, while both rs2111485 and rs13422767 are situated in the intergenic space, 13 kb and 23 kb apart from the end of the IFIH1 gene, respectively (Fig. 1). For all four SNPs, a major allele was related to increased diabetes risk, providing OR ranging from 1.7–2.3 (P = 6 × 10−4–8 × 10−8) in carriers homozygous for the major allele, compared to those homozygous for the minor allele (Table 1) (47). In Canada, two markers, rs2111485 A/G and rs984971A/G, both located in the intergenic regions, showed significant association with T1D, with OR for the minor allele G of 0.84 and 0.85, respectively (78). The lack of an association between the IFIH1 locus and T1D observed in Spanish (52) and Belgian (3) populations is likely explained by the limited power of these population analyses, particularly for the study by Martínez et al. (52), which involved only 311 affected and 535 control individuals. In addition, only a single marker of IFIH1 (rs1990760) has been evaluated in both studies.
Trio, a family consisting of two parents and one affected child.
The family dataset comprises multiplex and simplex Caucasian T1D families collected in the U.K., U.S.A., Ireland, Romania, and Norway.
The dataset includes families from the study of Smyth et al. (95), extended by additional Caucasian families from Europe and the U.S.A., and 228 families from the Asia-Pacific region.
The dataset consists of T1D nuclear families of Canadian Caucasians, with 40% from the province of Quebec.
The family dataset contains multiplex and simplex Caucasian T1D families from Europe and the U.S.A.
The dataset includes T1D nuclear families.
Abbreviations: IF1H1, interferon induced with helicase C domain 1; GCA, grancalcin; N/A, not available.
In the coding region of IFIH1, the non-synonymous substitution rs1990760 does not lie in any functional portion of the helicase molecule (Fig. 1), but the Ala946 allele is highly conserved among vertebrates (95). Recently, Shigemoto et al. (93) showed that this IFIH1 variant influences neither dsRNA binding nor IFN gene activation (Table 2). Another disease-associated marker, rs3747517, resides in the so-called MPH1 domain, which is conserved in ERCC4-like helicases and comprises two functional (helicase and C-terminal) subdomains (63). Liu et al. (47) found that rs1990760 affects the sequence of the putative binding site of the transcription factor HNF-3b, whereas rs3747517 alters the binding site for the transcription regulator AP-1. However, these SNPs are located in the coding region that is 45–50 kb from the start codon, and it remains to be determined whether these SNPs play a role in regulating IFIH1 expression.
Functional activity of SNPs was evaluated by Shigemoto et al. (93).
Abbreviations: IF1H1, interferon induced with helicase C domain 1; dsRNA, double stranded RNA; ND, not determined; OR, odds ratio; SNP, single nucleotide polymorphism; T1D, type 1 diabetes.
Liu et al. (47) reported a significant increase in basal levels of expression of MDA5 in peripheral blood mononuclear cells from the carriers of IFIH1 variants (i.e., homozygous for the major allele of rs3747517, rs1990760, rs2111485, and rs13422767), which was associated with a higher diabetes risk than those who had low-risk genotypes. Therefore, elevated expression of MDA5, which is likely to be upregulated after viral infections (5), could be implicated in the pathogenesis of T1D. These observations are in accordance with the recent results of Nejentsev et al. (62), who found an association between four rare variants (minor allele frequency <3%) of IFIH1 and T1D. Among those, the marker rs25667974 A>G (NP_071451.2:p.I923V) showed the most robust association (with the minor Val923 allele [OR = 0.51] in the case-control study, and 0.60 in the family-based study; Poverall = 2.1 × 10−16). This variation, which affects a highly conserved amino acid isoleucine at codon 923, is a loss-of-function mutation, since it significantly reduces IFIH1-dependent synthesis of IFN-β, but does not affect dsRNA binding (Table 2) (93).
Interestingly, other protective rare variants resulted in either the truncated protein (i.e., loss of the C-terminal 399 amino acid residues in the case of rs35744605 G>T [NP_071451.2:p.E627X]), or splicing disturbances caused by rs35337543 (NM_022168.2:c.1641+1G>C) and rs35732034 (NM_022168.2:c.2807+1G>A), which are located in introns 8 and 14 of IFIH1, respectively. Shigemoto et al. (93) showed that the truncated E627X product is non-functional, lacking both dsRNA binding and signaling activities (Table 2). The rare variants that disrupt IFIH1 function in the host antiviral response are likely to be negatively selected, rather than positively selected, since they confer protection from T1D.
The findings by Nejentsev et al. (62) suggest that rare genetic variants may have strong effects in complex diseases such as T1D. For IFIH1, the protective effects of these low-frequency variants on T1D risk (OR = 0.51–0.74) in U.K. Caucasians were even stronger then those of the common SNP rs1990760 (OR = 0.85) (95). Thus, the carriage of protective IFIH1 variants correlates with lower production of this helicase or its functional disruption, while upregulated MDA5 expression should promote the development of T1D in susceptible individuals. This hypothesis needs to be tested in future studies.
A growing body of recent epidemiological and immunological evidence suggests a role of enterovirus and picornavirus infections (whose antiviral response is mediated by IFIH1) as the sole agents in the development of T1D (37). Enteroviruses exhibit islet cell tropism, as demonstrated by the identification of expression of the coxsackievirus receptor by human islet endothelial cells (115), detection of viral RNA by in-situ hybridization, and finding viral proteins using immunohistochemical staining of post-mortem pancreatic specimens from T1D patients (21,112). Furthermore, an infectious coxsackievirus B4 was isolated from the islets of a type 1 diabetic patient (21), and a strain of echovirus 3 was isolated from an individual concurrently with appearance of islet cell and IA-2 autoantibodies (111). In accord with these findings, Richardson et al. (75) reported a high frequency of an enterovirus antigen in the autopsy pancreata of youth-onset type 1 diabetic patients by immunochemical detection of the enteroviral capsid vp-1 protein antigen in 44 of 72 diabetic subjects.
Interestingly, viral infections may not only induce, but also protect from T1D. In NOD mice, a murine model of autoimmune diabetes, infection with coxsackievirus and rotavirus has been shown to produce long-term protection from diabetes (31,90). However, the viral infection protects from diabetes when it precedes the inflammatory process (insulitis) in the exocrine pancreas. The degree of insulitis influences the timing of diabetes onset (23). In NOD mice with established insulitis, rotavirus infection was found to accelerate T1D (32). One possible mechanism by which an infection may trigger the development of T1D could result from the influence of the viral infection on the balance between IFN-γ and IL-4, two regulatory cytokines that promote or inhibit, respectively, the autoimmune process, leading to the development of T1D (90). Depending on the timing, a viral infection may alter the balance of IFN-γ or IL-4 production in the pancreas or elsewhere, causing a significantly divergent outcome in the initial progression toward diabetes.
A recent report provides evidence that signaling through cytoplasmic helicases/IFIH1 resulted in a markedly higher capacity of human monocyte-derived dendritic cells (DCs) to stimulate CD4+ T-cell proliferation (33). These results suggest that signaling through IFIH1 may result in immunogenic DCs, that may promote the development of diabetes. To evaluate the biological mechanism linking enterovirus infection with T1D, future functional experiments should test whether IFIH1-mediated immune activation caused by enterovirus infection may stimulate autoreactive T cells, leading to T1D, and whether suppressing IFIH1 can disrupt this pathogenic mechanism. It would also be interesting to evaluate whether the degree of the autoimmune and inflammatory response in the exocrine pancreas in T1D patients infected with an enterovirus correlates with levels of MDA5 expression, depending on the IFIH1 genotype.
It should be noted that the helicase gene lies on chromosome 2q24.3, within the linkage disequilibrium (LD) block that includes three more genes, encoding fibroblast activation protein-α (FAP-α), grancalcin (GCA), and a potassium voltage-gated channel and subfamily H member 7 (KCNH7) (Fig. 1). In addition to IFIH1, these three cannot be excluded from being candidates that harbor the etiological variant associated with T1D. The FAP-α gene product has been identified as a human stromal antigen, which is able to stimulate cytotoxic T-cell responses (25). The GCA gene product is abundant in neutrophils and macrophages, and is associated with degranulation and the consequent immune reaction (11). Thus, beside the antivirus-related type 1 diabetes mechanism of IFIH1, FAP-α and GCA may participate in the autoimmune or inflammatory destruction of pancreatic β cells. Finally, KCNH7 is also a functional candidate for T1D, as it may have a role in insulin secretion (59). Since genome-wide scans often do not reveal the precise causative sequence variants in such LD blocks, the FAP-α, GCA, and KCNH7 genes need further evaluation.
There is some evidence supporting a role of IFIH1 as the true etiological gene on chromosome 2q24.3. First, none of the SNPs located in the GCA, FAP-α, and KCNH7 genes showed an association with T1D in U.S. Caucasians (47) or Canadians (78). Second, Liu et al. (47) measured expression of both IFIH1 and GCA in peripheral blood mononuclear cells of T1D patients and non-diabetic controls, and observed significant genotype-expression correlation only for IFIH1, not for GCA. Finally, the identification of multiple rare T1D-associated variants in IFIH1 by Nejentsev et al. (62) points to its etiological role in T1D, because it is highly unlikely that multiple untested variants elsewhere in the region could explain the association of the rare IFIH1 variants of the LD. These findings strongly indicate a role for IFIH1 in the development of autoimmune diabetes.
IFIH1 and multiple sclerosis
Recently the IFIH1-GCA-KCNH7 genomic region was evaluated for association with MS in three Caucasian populations. In Spain, a significant association was found for rs2068330 (NM_033272.2:c2963-859G>C), located in intron 14 of KCNH7 (52). The minor allele G of rs2068330 was related to a reduced MS risk (OR = 0.73; p = 0.001). The marker rs1990760 of IFIH1 showed a trend toward association with MS in the Spanish population (for allele Thr, OR = 0.84; p = 0.07). In Denmark, for two non-synonymous SNPs, rs3747517 and rs1990760, located in the IFIH1 gene, a trend toward association with MS was observed (24). However, a family-based study in France failed to replicate this observed association for markers in the IFIH1 and KCNH7 genes (17). On the basis of these data, it is difficult to discern whether the IFIH1 or KCNH7 gene confers susceptibility to this organ-specific autoimmune pathology on chromosome 2q24.3. The KCNH7/ERG3 gene remains a strong positional candidate for MS. In the central nervous system, the gene product regulates neuronal excitability (73). Nevertheless, more large-scale and well-powered population studies are required to discover if there is a true MS-associated gene on chromosome 2q24.3.
Epidemiological studies have shown that enterovirus and cardiovirus infections seem to have no correlation with MS (14,18,44). However, in these studies only small cohorts of patients have been analyzed. Theiler's murine encephalomyelitis virus (TMEV) and related theiloviruses, which are specifically recognized by MDA5, cause encephalomyelitis in rodents and humans, a disease with a persistent infection of the central nervous system, and inflammatory immune-mediated demyelination that is very similar to MS (109). Experimental data obtained from mice with TMEV-induced encephalomyelopathy, an animal model of MS, support the possibility of a virus-induced autoimmune response against myelin mediated by self-reactive T cells (57,65,66). The development of viral-induced autoimmune demyelinating disease is accompanied by induction of the innate immune response, which suggests a putative role of IFIH1 in the pathogenesis of MS (67). However, the precise role of this virus-sensing helicase in the pathophysiology of MS remains to be identified and further characterized.
IFIH1 and other autoimmune disorders
The IFIH1 gene was tested for association with several other autoimmune diseases, including GD, RA, and autoimmune Addison's disease (AAD). In a British population, Sutherland et al. (97) studied 204 AAD patients and 446 controls, but observed no association between rs1990760 and AAD. These researchers found a strong association between this SNP and GD after the analysis of 602 affected patients. The Ala946 allele of rs1990760 contributed to a higher GD risk (OR = 1.47; p = 1.9 × 10−5). However, Zhao et al. (116) failed to confirm the association between rs1990760 and GD in a Chinese population consisting of 261 GD subjects and 206 non-affected individuals. This lack of association in the Chinese may result from ethnic differences, since the Thr946 allele, which is minor in Caucasians, is dominant in the Chinese (Ala vs. Thr = 0.57/0.43).
There are no established links between GD and viral infection (104). The finding of a strong association of IFIH1 alleles with GD in British Caucasians may suggest that IFIH1 has an endogenous or indirect immunoregulatory effect, which is unrelated to any specific role as a viral receptor. Further replication studies are required to understand whether IFIH1 plays a role in autoimmune thyroid disease.
Recently, the association between helicase gene variants and RA was evaluated in two reports. In the U.K. Caucasian cohort (965 affected and 988 healthy individuals), no significant association was found between the marker rs1990760 and RA (51). However, a Spanish study involving 540 RA patients and 535 controls showed a trend toward association with this marker (OR for allele Thr = 0.85; p = 0.058), and a moderate association with RA for the intronic polymorphism rs2068330 (NM_033272.2:c.2963-859G>C), located in the neighboring KCNH7 (53). The minor allele G of rs2068330 was related to a decreased risk of RA (OR = 0.8; p = 0.016). These data provide evidence that the IFIH1-GCA-KCNH7 region harbors an etiological marker for RA that remains to be discovered. Additionally, Marinou et al. (51) observed significantly greater levels of IFIH1 expression in RA patients than in controls. Although these investigators did not report whether the IFIH1 genotypes correlated with MDA5 expression, their findings are consistent with the observations of Liu et al. (47), suggesting the viral upregulation of MDA5 expression as a likely mechanism involving IFIH1 in the induction of autoimmunity.
A possible role of viruses such as Epstein-Barr virus, parvovirus B19, human herpesvirus-6, cytomegalovirus, and others, in the pathogenesis of RA has been suggested (15,27,77). However, there are no publicly available clinical reports showing the presence of coxsackievirus and other picornaviruses, which are specific targets for recognition by MDA5, in the pathogenesis of RA. It cannot be excluded that, regardless of virus infection, MDA5 expression may be secondarily stimulated in RA patients by IFN-β, whose overexpression was demonstrated in rheumatic sinovial tissue (107). The increased expression of IFN-β may serve as a biomarker of the induction of a defensive immunomodulatory mechanism that could inhibit synovial inflammation (98).
Other factors in the antiviral immune signaling mediated by RIG-I-like helicases: Genetic implications for autoimmunity
Except for MDA-5, no other inducers of the innate immune signaling regulated by RIG-I-like RNA sensors have been studied for associations with autoimmune and inflammatory diseases. Potter et al. (74) failed to show an association between the TRAF3 gene and RA in British Caucasians. For IRF3, two promoter polymorphisms, −925A/G and −776C/T, showed an association with systemic lupus erythematosus (SLE) in a small Japanese population sample (1). Patients homozygous for the minor G-T/G-T haplotype of IRF3 had a decreased risk of SLE, and significantly lower IRF3 mRNA expression. However, the association between IRF3 and SLE was not replicated in a much larger Spanish cohort (85). For caspase-8 and caspase-10, whose role in the genetic susceptibility to cancer is well established, a modest association of rs6750157 in caspase-10 and rs1045485 in caspase-8 with asthma was shown in U.S. African-Americans and U.S. Hispanics (94). More genetic studies should be performed to reveal whether sequence variations in the members of RIG-I and MDA5-dependent signaling significantly contribute to autoimmunity.
Conclusions
The results of recent genetic and functional studies support a likely role of IFIH1 in the pathogenesis of T1D. There is preliminary evidence for the implication of this helicase in several other autoimmune diseases, such as GD, RA, and MS. However, this requires further substantial efforts to confirm or reject the contribution of the IFIH1 gene to susceptibility to these disorders. Thus at present, it is too early to consider IFIH1 as a general autoimmunity gene that links viral infections to autoimmunity. Although the molecular mechanisms by which MDA5 is involved in the autoimmune response are not yet fully understood, it is clear that IFIH1 gene variants associated with low MDA expression, or those abolishing the function of this helicase, could be related to a reduced risk of autoimmunity. The discovery by Nejentsev et al. (62) of several rare IFIH1 variants that account for T1D susceptibility on chromosome 2q24.3 through their effects on MDA5 function and expression suggest a novel pathogenic pathway for T1D. Such a finding is not unique, since for example, Vinuesa and Cook (108) characterized a genuine effect of a mutation at the Roquin gene on the pathogenesis of SLE-like autoimmune disease in N-ethyl-N-nitrosourea-mutagenized mice. To date, the contribution of rare genetic variants to autoimmunity remains unclear, and thus has yet to be extensively evaluated and understood. For those rare IFIH1 variants that demonstrate apparent associations with T1D, future studies are needed to determine whether these alleles are associated with other autoimmune diseases.
The elevated levels of IFIH1 observed in patients with T1D and RA suggest the upregulation of the expression of this helicase. The mechanism that causes the stimulation of MDA5 expression in autoimmunity remains to be elucidated. There is likely a causative role of viruses in the upregulation of IFIH1 in autoimmunity. However, whether viral infections are a major cause of the activation of MDA5 expression in autoimmunity remains to be clarified.
As documented in several reports, coxsackievirus was found in patients with primary Sjögren's syndrome (SS) (105,106). Levels of SS-specific autoantibodies in a general population were shown to correlate significantly with levels of enterovirus antibodies (55). Based on these data, Triantafyllopoulou and Moutsopoulos (105) suggested a role of persistent virus infection as a trigger in inducing SS in genetically prone individuals. Since coxsackievirus belongs to the family of picornaviruses specifically recognized by MDA5, IFIH1 may represent a causal link between the enteroviral infection and genetic predisposition to SS. In the future, it would be valuable to determine whether IFIH1 variants are implicated in the development of SS.
The molecular pathways linking upregulated expression of IFIH1 with increased risk of autoimmunity require further study. Finding a role for IFIH1 in increasing the ability of DCs to activate proliferation of CD4+ T cells (33) may be one such link. This observation is supported by the findings of Longhi et al. (48), who observed that MDA5 is essential for poly I:C-RNA-induced production of type I IFNs by DCs and monocytes against DCs targeted with HIV gag protein vaccine in mice. Type I IFNs are required for DC maturation and the development of CD4+ T-cell-mediated immunity and autoimmunity (102).
In further support of the likely involvement of IFN-α in mediating virus-upregulated expression of IFIH1 and autoimmunity, the ability of poly-I:C, a mimic of viral dsRNA, to induce insulitis and autoimmune diabetes in animal models has been reported in several studies (19,20,58,71). As shown by numerous clinical case reports, treatment with IFN-α is often associated with the development of autoimmunity (89). Elevated serum levels of IFN-α were also detected in autoimmunity (10,12,45).
Virus-stimulated expression of IFIH1 may lead to increased expression of IFN-α. IFN-α, in turn, could contribute to autoimmunity via several mechanisms (Fig. 3). This interferon could have direct cytotoxic effects on target cells via stimulation of DCs, or cause apoptosis and necrosis of infected cells through activation of the oligoadenylate synthetase/RNAse L pathway (84), or via RNA-dependent protein kinase/NFκβ-dependent caspase activation (54). The release of apoptotic materials may then induce the autoimmune response and stimulate plasmacytoid dendritic cells (PDCs, or immature DCs) to further produce IFN-α, that could further enhance self-immune reactivity. In patients with primary SS, RNA-containing immune complexes were shown to trigger the ability of PDCs to prolong IFN-α synthesis, promoting a cyclical mechanism of autoimmune exacerbation via increased autoantibody production, and the formation of more endogenous IFN-α inducers (10).
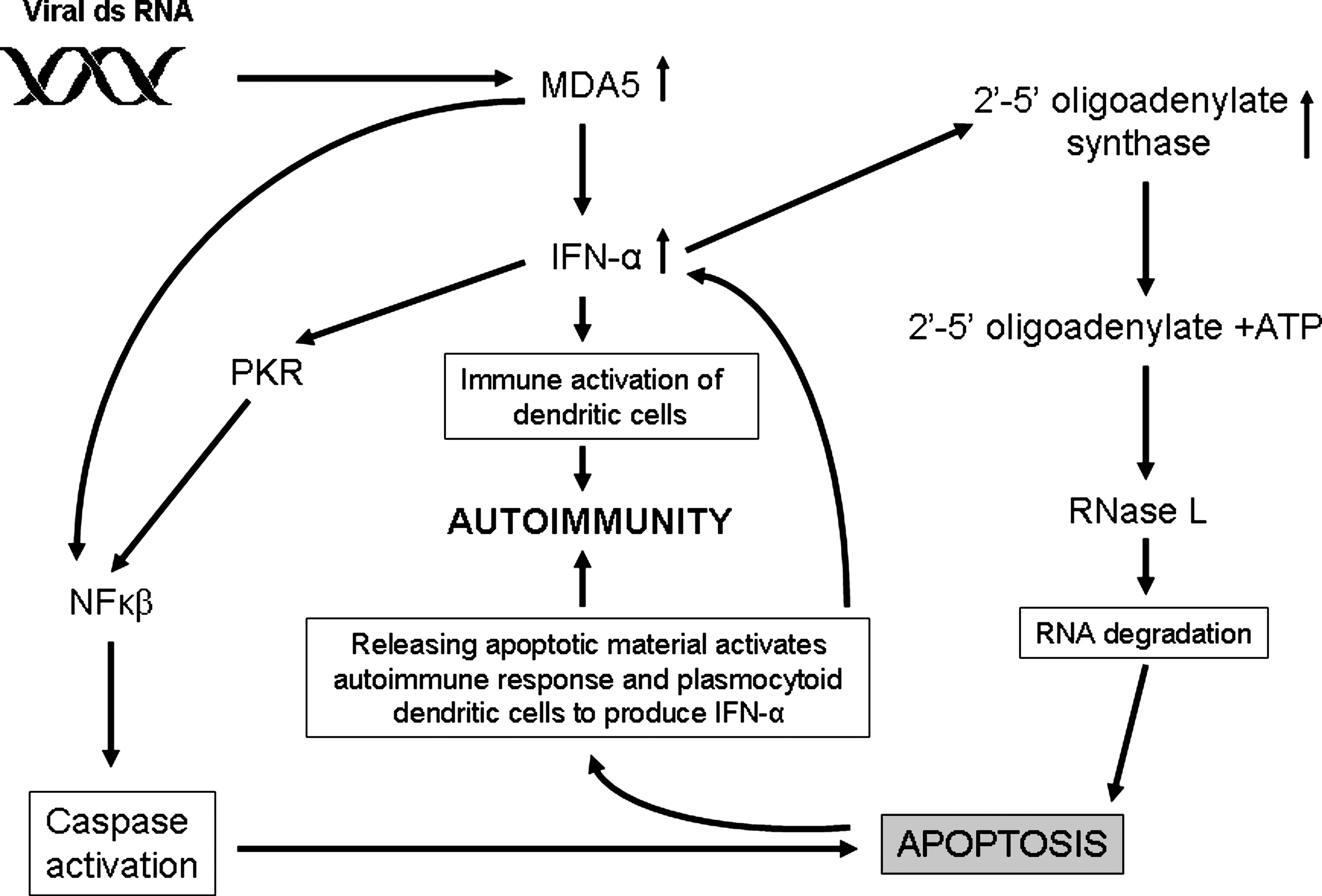
Interferon-α as a putative link between virus-induced upregulation of IFIH1 and autoimmunity. Viral dsRNA activates expression of MDA5, a product of the IFIH1 gene. In viral infection, MDA5 expression is frequently upregulated and may lead to overexpression of IFN-α. Activation of IFN-α, in turn, stimulates dendritic cells and other immune cells, which are directly cytotoxic to target cells infected with viruses. IFN-α could induce cell apoptosis by several possible mechanisms, including activation of the oligoadenylate synthase (OAS)-RNase L pathway, and the protein kinase R (PKR)-dependent pathway. The first pathway blocks viral replication by mRNA degradation and leads to cell death and destruction. PKR induces nuclear factor-κβ (NF-κβ)-mediated caspase activation and apoptosis. Apoptotic materials induce more IFN-α and activate the immune system. This could promote and exacerbate a cyclical mechanism of autoimmune induction.
Footnotes
Author Disclosure Statement
No competing financial interests exist.