
Abstract
Previously, we showed that intraperitoneal infection with murine coronavirus strain JHM (JHMV) established a persistent infection with subacute granulomatous serositis in interferon-γ-deficient C57BL/6 (B6-GKO) mice. Herein, we characterize a variant virus from B6-GKO mice persistently infected with JHMV. Viruses were isolated from ascites at 25 d post-infection and cloned by limiting dilution on DBT cells; one variant was named 25V16G. To compare pathogenicity in vivo, we inoculated 25V16G and JHMV intraperitoneally into 8- to 12-week-old B6-GKO mice. Whereas nearly all of the B6-GKO mice infected with JHMV survived over 14 d, all of those infected with 25V16G died by 9 d post-infection. Histopathological examination revealed that 25V16G induced acute fulminant hepatitis in B6-GKO mice, whereas JHMV caused severe but focal hepatitis. The virus titer of 25V16G in the liver was 50- and 250-fold higher than that of JHMV at 5 and 7 d post-infection, respectively. However, there was no significant difference in viral growth between 25V16G and JHMV in cell lines cultured in vitro. Nucleotide sequencing of the S gene of 25V16G and JHMV revealed a deletion of 29 amino acids encompassing S511–539, which covers a major cytotoxic T lymphocyte (CTL) epitope in C57BL/6 mice, and two point mutations resulting in amino acid changes in the S protein of 25V16G. One explanation for the greater pathogenicity of 25V16G is that 25V16G escapes CTL-mediated protection in B6-GKO mice. This experimental model may be used to assess the role of IFN-γ in viral persistence in vivo.
Introduction
Murine coronaviruses induce a variety of diseases in mice, including hepatitis, enteritis, and encephalitis. Such diseases depend on the virus strain, infection route, age, genetic background, and immune status of the host (4,19). Previously, we reported reduced viral clearance and resultant systemic persistent infection associated with disseminated granulomatous serositis in interferon-γ (IFN-γ)-deficient C57BL/6 (B6-GKO) mice after intraperitoneal (IP) infection with murine coronavirus strain JHM (JHMV; 21). The same disease has also been observed in IFN-γ-deficient B6 mice naturally infected with murine coronavirus (10). However, the mechanism of viral persistence in B6-GKO mice after murine coronavirus infection remains unknown. Moreover, it has unraveled how a virus can induce granulomatous serositis, which is similar to a distinctive lesion of feline infectious peritonitis, a coronavirus-induced fatal disease in cats (2).
In this study, variant viruses were isolated from a persistently infected B6-GKO mouse and characterized. One variant virus, named 25V16G, was more virulent than the parental JHMV in B6-GKO mice. One explanation for the increased pathogenicity of 25V16G is that 25V16G escapes cytotoxic T lymphocyte (CTL)-mediated protection in B6-GKO mice. This experimental model may be used to assess the role of IFN-γ in viral persistence in vivo.
Materials and Methods
Mice
IFN-γ-deficient mice of C57BL/6 background (B6-GKO) were obtained by back-crossing a 129/SvJ mouse with a disrupted IFN-γ gene (29) with C57BL/6 mice more than 10 times. The genotype of the mice was determined by PCR as described previously (20), and 8- to 12-week-old female mice were used. Breeding mice were maintained in a laminar-flow rack in an environmentally controlled area and checked routinely to ensure that they were serologically free of murine coronaviruses and other pathogenic agents. The mice were infected IP with 1 × 106 PFU of JHMV in a volume of 0.2 mL. The infected mice were kept in a safety cabinet in a different area. The experiments were conducted in accordance with the University of Tokyo Institutional Guidelines for Animal Experiments, and Safety Guidelines for Gene Manipulation Experiments.
Viruses and cells
The DL variant of JHMV was propagated and plaque assayed on DBT cells as described previously (20). IC-21 cells and J774A.1 cells were cultured as described previously (21,32). The IC-21 and J774A.1 cells were macrophage-like tumor cell lines derived from C57BL/6 and BALB/c mice, respectively. For viral titration, 10% tissue homogenates of samples were serially diluted and plaque assayed on DBT cells (20). For viral isolation, serially diluted samples were inoculated onto DBT cells. For virus cloning, limiting dilution was performed using DBT cells cultured in 96-well plates.
Histopathology
Tissue samples were fixed in 10% phosphate-buffered formalin, embedded in paraffin, sectioned, and stained with hematoxylin and eosin.
RT-PCR
Total RNA from DBT cells infected with either 25V16G or JHMV was extracted using the ISOGEN method according to the manufacturer's instructions. cDNA was synthesized from 1 μg total RNA using the Reverse Transcription System (Promega Corp., Madison, WI) and oligo(dT) primers according to the manufacturer's instructions. PCR was performed as described elsewhere (1). Briefly, the primer sequences used were as follows: SI-1, 5′-TAT GAA TTC TAC GTT ATG TCC AGG CTG AGT C-3′; and SI-2, 5′-TAT GGA TCC ATA GAG GTC ATA TCT GAC GC-3′. The reaction mixture was incubated at 94°C for 5 min, and then subjected to 30 cycles of 94°C for 1 min, 60°C for 1 min, and 72°C for 1 min, followed by a final extension step at 72°C for 7 min. PCR products were resolved by electrophoresis and stained with ethidium bromide.
cDNA cloning and sequencing
Total RNA from DBT cells infected with either 25V16G or JHMV was extracted using proteinase K, phenol, and chloroform. Poly(A)-tailed RNA was selected using an oligo(dT) column (Collaborative Research Inc., Bedford, MA). The cDNA cloning and sequencing of the S gene were performed as previously described (30).
Statistical analysis
Data were assessed for statistical significance using Student's t-test. Differences were considered to be statistically significant at p < 0.05.
Results
Isolation of 25V16G from B6-GKO mice persistently infected with JHMV
B6-GKO mice were inoculated with JHMV. At 25 d post-infection, ascitic fluid was removed from mice with conspicuous abdominal distension due to ascites formation. The fluid was inoculated onto DBT cells. After overnight incubation, syncytium formation, an apparent cytopathic effect (CPE) of MHV, was observed, and the culture supernatant was removed. The virus was cloned by limiting dilution three times based on virus-induced CPE. We obtained 10 clones, and one clone, designated 25V16G, was analyzed in this study. There was no significant difference in plaque size or morphology between 25V16G and JHMV (data not shown).
Acute fulminant hepatitis in B6-GKO mice after IP infection with 25V16G
To examine the pathogenicity of 25V16 in B6-GKO mice, we inoculated 8- to 12-week-old B6-GKO mice IP with 1 × 106 PFU 25V16G or parental JHMV and monitored them for 2 wk (Fig. 1). Whereas nearly all of the B6-GKO mice infected with JHMV survived over 14 d in agreement with previous results (20,21), all of those infected with 25V16G died by 9 d post-infection. These results clearly indicate that the pathogenicity of 25V16G in B6-GKO mice is greater than that of the parental JHMV.
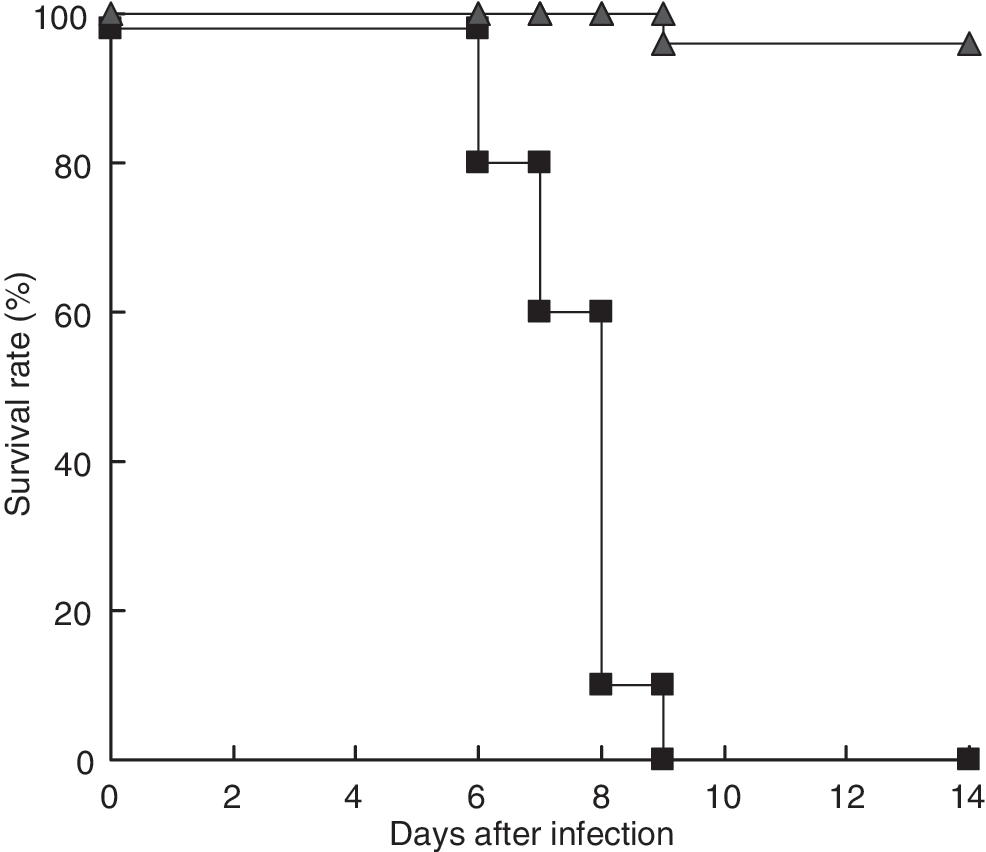
Survival rate of B6-GKO mice after IP infection with 25V16G or parental JHMV. B6-GKO mice (n = 24 and 10) were inoculated IP with 1 × 106 PFU 25V16G (▪) and JHMV (▴), respectively, and monitored for 2 wk.
To understand the pathological changes in virus-infected B6-GKO mice, we compared histopathological changes of the liver in B6-GKO mice infected with 25V16G and JHMV (Fig. 2). As reported previously (20,21), JHMV induced necroinflammatory foci containing mostly mononuclear cells in the livers of B6-GKO mice at 7 d post-infection (Fig. 2A and C). In contrast, massive necrosis with mononuclear cell infiltration and severe hemorrhage was observed in 25V16G-infected B6-GKO mice at 7 d post-infection (Fig. 2B and D), which was clearly different from the focal necrotic lesions observed in JHMV-infected B6-GKO mice. No other serious lesions were observed in 25V16G-infected B6-GKO mice (data not shown).
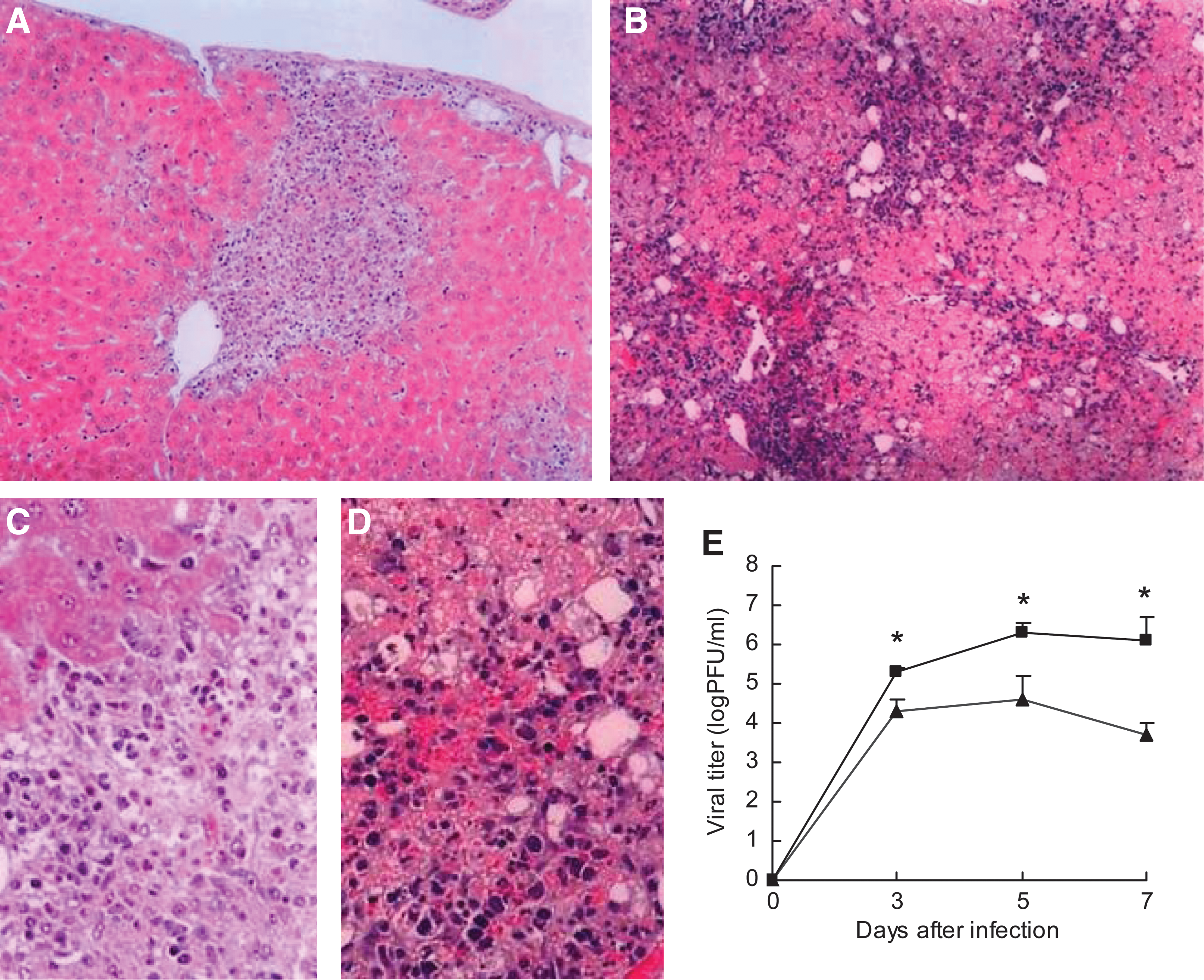
Histopathological changes in the livers of B6-GKO mice after IP infection with 25V16G and parental JHMV. Shown are the pathological changes in the liver from (
To assess the viral burden, we determined the viral titers of the liver from 25V16G- or JHMV-infected B6-GKO mice. At 3 d post-infection, the viral titer in the livers of 25V16G-infected B6-GKO mice was significantly higher than that in the liver of parental JHMV-infected B6-GKO mice (Fig. 2E). Similarly, viral titers of 25V16G-infected B6-GKO mice were 50- and 250-fold higher than those of JHMV-infected B6-GKO mice at 5 and 7 d post-infection, respectively. Taken together, these results indicate that the stronger virulence of 25V16G in B6-GKO mice is associated with a higher viral burden in the liver.
Growth potential of 25V16G is not superior to JHMV in cultured cell lines
To assess the replication efficiency of 25V16G, we compared the viral growth of 25V16G and JHMV in three cultured cell lines (Fig. 3). DBT, IC-21, and J774A.1 cells were inoculated with either 25V16G or JHMV, and the viral titer of the culture supernatant was determined by plaque assay. As shown in Fig. 3, there was no significant difference in viral titer between 25V16G and JHMV in the three cell lines tested. There was no difference in the CPE between the two viruses in the three cell lines (data not shown). These results suggest that the replication potential of 25V16G is comparable to that of parental JHMV. A difference in viral titer might be observed only in B6-GKO mice.

Replication of 25V16G and parental JHMV in cultured cell lines. (
Sequence analysis of the S gene of 25V16G
Because the S protein has many important biological functions (9,15,22,24,27), the nucleotide sequence of the S gene was compared between 25V16G and JHMV. Before sequencing, we performed RT-PCR analysis of the S gene (Fig. 4A), and observed that the RT-PCR product from 25V16G appeared to be smaller than that from the wild-type JHMV, suggesting that 25V16G has some deletion in the S gene. The RT-PCR products obtained from the other nine variants were also shorter than that of the wild-type JHMV, although some variation was observed among clones (Kyuwa et al., unpublished data). Finally, the nucleotide sequence analysis revealed that 25V16G had a deletion of 29 amino acids (S511–539) as expected, as well as two point mutations causing substitutions (C490S and G771V) in the S protein (Fig. 4B).
Comparison of the S gene between 25V16G and parental JHMV. (
Discussion
Viral persistence is a major topic in virology. Many diseases are caused by chronic virus infections, for example AIDS caused by HIV, chronic hepatitis and hepatocellular carcinoma caused by infection with hepatitis B and C viruses, cervical cancer caused by human papillomavirus, and subacute sclerosing panencephalitis caused by measles virus (6,7,12,14). To develop safe and affordable methods and strategies for combating chronic virus infections, it is necessary to further improve our knowledge of viral persistence.
Previously, we reported disseminated granulomatous serositis in B6-GKO mice associated with persistent murine coronavirus infection (21). To better understand the mechanism of viral persistence in B6-GKO mice after murine coronavirus infection, as well as the etiology of granulomatous serositis, in the current study we characterized a variant virus, 25V16G, which was isolated from ascitic fluid at the subacute phase. Our results demonstrate that 25V16G is more virulent than the parental JHMV in B6-GKO mice, and causes a higher viral burden. In contrast, our experiments in an in vitro culture system using three cultured cell lines indicated that 25V16G does not surpass the parental JHMV replication. There are three possible explanations for this. The first hypothesis is that 25V16G is a hepatocyte-adapted variant that replicates well only in hepatocytes; the second is that the in vivo tropism of 25V16G is changed and wider than that of wild-type virus; and the third is that 25V16G and JHMV have the same potential to replicate, but only 25V16G has the potential to evade immune surveillance in vivo.
CTLs play an essential role in virus clearance in vivo (5,28). In JHMV infection, residues 510–518 of the S protein (S510–518) comprise the immunodominant CTL epitope in C57BL/6 mice (3,27). The importance of CTL-mediated viral clearance in JHMV infection has been demonstrated by the various mutations observed in the CTL epitope in viruses that persist in vivo (25). Moreover, CTL escape mutants help cause JHMV-induced demyelinating disease (27). Finally, Pewe et al. have shown that infection with CTL escape mutants results in increased mortality (26).
Although the same virus (JHM strain) was used in this study, our experimental model was totally different. For example, Pewe et al. established a persistent infection in the central nervous system of suckling B6 mice that had been infected intranasally with JHMV at 10 d of age and nursed by dams previously immunized against the virus (27). In our model, a systemic persistent infection was established in young adult B6-GKO mice by IP infection with JHMV (21). Because the balance between viruses and the host's immune response is critical in establishing a persistent infection, a host must mount partially protective antiviral immune responses in each model.
In this study, we found that 25V16G has a deletion of 29 amino acids encompassing S511–539, which covers a major CTL epitope in C57BL/6 mice, and two point mutations leading to amino acid changes in the S protein. Excellent studies on the mechanism of persistent JHMV infection in the central nervous system (3,25 –28) have led us to one likely explanation, that 25V16G is a variant that escapes CTL-mediated protection in B6-GKO mice. Of course, further studies are needed to elucidate the detailed mechanism of a systemic persistent JHMV infection in B6-GKO mice. First, we must address the other two hypotheses mentioned above. We cannot exclude the possibility that the two point mutations (C490S and G771V), and other unidentified mutations other than S gene, play some role in the increased virulence in B6-GKO mice. We are also interested in the specificity and role of antiviral antibodies detected in persistently infected B6-GKO mice (21). In any case, our experimental model may be useful for assessing the role of IFN-γ in viral persistence in vivo.
Conclusion
In this study, we characterized a variant virus from B6-GKO mice persistently infected with JHMV. The variant, designated 25V16G, displayed higher virulence than the parental strain in B6-GKO mice, and was associated with a higher viral burden in the liver. However, there was no significant difference in viral growth in cell lines cultured in vitro. We identified a deletion of 29 amino acids encompassing S511–539, which covers a major CTL epitope in C57BL/6 mice, and two point mutations encoding amino acid changes in the S protein. One explanation for the increased pathogenicity of 25V16G over the parental strain is that 25V16G escapes CTL-mediated protection in B6-GKO mice.
Footnotes
Acknowledgments
We thank Ms. H. Kubota for technical assistance. This work was supported in part by a grant-in-aid from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Author Disclosure Statement
No competing financial interests exist.