
Abstract
Infection with human papillomaviruses (mostly HPV6 and HPV11) may lead to recurrent respiratory papillomatosis (RRP), a chronic disease affecting 2–4/100,000 people. Papillomas have to be removed surgically so patients can breathe normally. Papillomas often grow back and some patients are subjected to a number of operations. In general, asymptomatic HPV-positive people have low levels of antiviral antibodies in their sera, as the human humoral response is weak due to HPV's biology. In patients suffering from RRP who have undergone multiple surgeries, a blood–epithelium barrier breach stimulates the production of anti-HPV antibodies. Our study's aim was to produce HisTag-HPV11-L1 major capsid protein in E. coli cells, and to purify it. We also sought to detect anti-HPV11-L1 antibodies in antisera obtained from RRP patients using ELISA. Clinical samples were collected from 47 patients with RRP (antisera and papillomas), and from 32 controls (sera and oral swabs), from the Wielkopolska region of Poland. Antisera and control sera were used to coat microplates, HisTag-HPV11-L1 antigen was applied, and antibody-antigen complexes were detected by anti-HisTag monoclonal antibody in an ELISA assay. Simultaneously, total cellular DNA was extracted from papillomas and oral squamous cells obtained from controls. All DNA samples were screened for HPV DNA using MY-PCR. All patients were HPV-positive (30% for HPV6 and 70% for HPV11). Statistically significant correlations were found between the amount of anti-HPV11-L1 antibodies in the sera of RRP patients and the number of surgical procedures they underwent. Although HPV virus-like particles are most often used for anti-HPV antibody detection, the ELISA method presented herein is another viable option for use in RRP patients.
Introduction
Two distinct forms of RRP have been described: juvenile-onset recurrent respiratory papillomatosis (JORRP), and adult-onset recurrent respiratory papillomatosis (AORRP). The HPV child transmission patterns include perinatal vertical transmission (30), skin-to-skin contact, and sexual abuse. JORRP is more aggressive and affects 4 out of 100,000 people. The most common treatments for RRP are surgical removal of papillomas with a CO2 laser, and the use of interferon or antivirals like cidofovir to slow the growth of papillomas (24,27). In more serious cases tracheostomy is necessary. There is hope that RRP may someday be prevented by vaccines (36).
The HPV-L1 capsid protein is a major papillomavirus immunogen (11). Previous infection with HPV does not assure immunity against future infections, because of the lack of a viremic phase in HPV-infected individuals (36). In general, the immune response against HPV infection is weak, due to the biology of HPV reproduction (which is limited to differentiating epithelial cells), but nevertheless cytokines are produced (TNF-α, IL-1, TGF-β, and IFN), viral antigens are presented on antigen-presenting cells (APCs) (11,17,29), and small amounts of IgG, IgA, and IgM antibodies are detected in serum (10).
In most HPV-infected people the virus is invisible to the hematologic system because the viral reproduction cycle is independent of blood cells; however, the presence of the HPV16 genome has been detected in peripheral blood mononuclear cells, but it is unknown if viral antigens and HPV virions are produced in these cells (2). Therefore the level of antibodies in serum may be useful as a marker of disease progression in those with HPV infection (15), particularly in RRP patients in whom the blood–epithelium barrier is breached during surgery. It may cause a serostatus similar to that of people vaccinated against HPV. Anti-HPV vaccines are produced using HPV-L1 protein that is synthesized primarily in eukaryotic cells (e.g., insect and yeast cells), in which the major capsid protein spontaneously forms highly immunogenic virus-like particles (VLPs) (35).
The aim of our research was to detect antibodies directed against HPV11-L1 protein in antisera from patients with JORRP using an enzyme-linked immunosorbent assay (ELISA). Elaboration of a method of evaluating the humoral response in JORRP patients was our secondary goal. In the approach presented herein, eukaryotic cells in which HPV11-L1 VLPs are usually produced (29,37) were replaced by E. coli cells, in order to synthesize the HPV11-L1 major capsid protein fused with a polyhistidine tag (HisTag) to simplify the preparation of viral antigen. There are recent reports of similar assays, in which bacterial cells were utilized for the synthesis of HPV6, 16, and 18 antigens, and the use of HPV16-L1 protein tagged with GST for validation of HPV serology (31,32). In other studies efforts were made to induce L1 protein purified from prokaryotic cells to assemble into VLPs in vitro (4,26). Escherichia coli was also utilized to produce pentameric L1 fusion protein as an economical alternative to VLPs in an anti-HPV vaccine, and it was shown that capsomeric/pentameric forms of L1 protein could induce protective immunity in the host (41). Also, HPV antigens produced in E. coli cells were used in denatured form in ELISA assays (7).
The principal goal of our study was to verify that HPV-11 L1 protein obtained from prokaryotic cells, without determination of its conformational state, can be recognized by human antibodies produced in patients with JORRP.
Materials and Methods
Study sample
Clinical samples (antisera and papillomas) were collected over a period of years (2006–2008) from 47 patients with JORRP. The youngest hospitalized child was 34 mo old, and the oldest was 12 y old. The first symptom was usually a phonation disorder, and in 13 cases the symptom was dysphonia. The time interval between the occurrence of the first symptoms and diagnosis ranged from 2–32 mo (average 8 mo) before hospital admission. Three of the children had undergone tracheostomy before hospitalization, two of them had a tube for 2 y before admission, and in one case tracheostomy had been performed 1 mo before hospitalization. The area affected with papillomatous changes was the glottis in 16 cases, the laryngeal vestibule in 11 cases, the glottis together with the subglottic region in 8 cases, and all levels of the larynx in 4 cases. The basic therapy performed was microsurgery to remove papillomatous growths in the larynx using forceps and Kleinsasser's directoscope, under microscopic magnification. The number of microsurgeries performed in the JORRP patients ranged from 1–39 (median 8). The recurrence of symptoms in many cases was manifested as spread of the papillomatous changes into formerly unaffected areas of the larynx and outside the larynx, namely to the trachea and bronchi (9 cases), the lower pharynx (4 cases), the middle pharynx and oral cavity (5 cases), and to the nose (7 cases). After each operation the resected tissue was subjected to histopathological examination.
Thirty-two control samples (sera and oral squamous cells) were collected from patients admitted for treatment at the Department of Pediatric Otolaryngology who were not suffering from JORRP. All patients and controls resided in the Wielkopolska region of Poland.
About 10–12 samples were received at our laboratory every few months, from both control individuals and JORRP patients. Oral squamous cells, papillomas, antisera, and sera from all studied subjects (n = 79) were transported frozen in a portable freezer from the hospital, and upon arrival at our laboratory all samples were stored at −20°C. All analyses (PCRs and ELISAs) were performed on each new series of samples within several days after arrival, and were repeated at the end of the study.
Sera and antisera were used to coat ELISA plates and HPV11-L1 protein was applied as an antigen. Sera and oral squamous cells from the control group were subjected to extraction of cellular DNA.
DNA extraction
Oral squamous cells from all control subjects (n = 32) were swabbed with a cotton-tipped applicator from different parts of the oral cavity and oropharynx. Total DNA isolation from oral squamous cells was performed with a ready-to-use set, the A&A Biotechnology Swab (cat. no. 025-100; A&A Biotechnology, Gdynia, Poland). About 20–50 mg of papillomatous tissue was submerged in liquid nitrogen, crushed with a mortar and pestle, then total DNA isolation from papillomas was carried out using a DNA tissue kit (Qiagen, Hamburg, Germany). Before further analysis, the quality of the cellular DNA was verified by PCR amplification of a fragment of c-fos, which is a cellular proto-oncogene present in each human cell (data not shown). This step was performed in order to confirm the presence of nucleated cells in oral swabs, and to validate the DNA extraction procedure, both from oral squamous cells and papillomas, assuming that the PCR product will be synthesized only if the isolated total DNA template is of good quality. All DNA isolates were measured in a spectrophotometer (λ = 260 nm), and brought to the same concentration for PCR analysis (∼20 ng/μL).
HPV-11 and HPV-6 genomic DNA detection
Total DNA extracted from oral squamous cells and papillomas was used as a template in MY-PCR amplification with MY09 and MY11 degenerate oligonucleotides complementary to the HPV-L1 open reading frame, allowing for detection of 33 HPV genotypes as previously described (28,22). All MY-PCR reactions were performed in the Biometra T-gradient thermocycler (Biometra GmbH, Goettingen, Germany) using the following procedure: (1) incubation with UNG at 55°C for 2 min; (2) pre-denaturing at 95°C for 5 min; (3) annealing of oligonucleotides at 55°C for 30 sec; (4) elongation at 72°C for 45 sec; (5) denaturing at 95°C for 30 sec; (6) final elongation at 72°C for 7 min. Steps 3–5 were repeated 40 times. Reaction volume was 10 μL, containing 1 μM of each oligonucleotide, 0.6 mM MgCl2, 1× KCl buffer for Taq polymerase, 0.4 U of Taq polymerase (Fermentas International, Burlington, Ontario, Canada), 0.1 U of UNG (Jena Bioscience, Jena, Germany), and 2 mM of each dNTP (dATP, dTTP, dGTP, dCTP, and dUTP). For diagnostic MY-PCR 20–50 ng of total DNA from all individuals (2 μL) were used, and 5 μL of MY-PCR products were electrophoresed in 1.5% agarose gel (Prona Agarose, Madrid, Spain) containing ethidium bromide. All positive PCR products containing 450-bp HPV PCR products were subjected to direct DNA sequencing in order to determine HPV genotype. MY-PCR samples containing visible bands were directly sequenced using 1–2 μL of the MY-PCR reaction, and 50 pmol of the sequencing oligonucleotide (both MY09 and MY11 in two separate sequencing procedures). Sequencing was performed with BigDye Terminator v3.1 on an ABI Prism 3130XL Analyzer (Applied Biosystems, Foster City, CA). Sequence chromatograms were checked for accuracy, and contigs were edited and assembled using FinchTV 1.3.1 (Geospiza Inc., Seattle, WA), and GenDoc 2.7.000. The nucleotide sequences were compared with GenBank sequences using the BLAST program.
In each MY-PCR round an appropriate MY-PCR control was processed concurrently (1): a positive control, a plasmid containing the HPV11-L1 open reading frame produced for our prior studies (8); and two plasmids containing HPV-L1 open reading frames from HPV16 and HPV18 (kindly provided by G. Zur Hausen and Marco Ciotti, respectively) were combined in equimolar proportion at an approximate final plasmid DNA concentration of ∼1 pg/μL. Then human cellular HPV-negative DNA (∼20 ng/μL) was added to the mixture of plasmids in a 1:1 volume ratio, and 2 μL was used as a positive control. Negative controls were water and/or HPV-negative samples.
Strict precautions were taken in order to avoid false-positive results, and good laboratory practice was implemented: (1) one-direction sample processing using separate rooms for DNA isolation, preparation of MY-PCR, thermal cycling, and electrophoresis; (2) UNG and dUTP addition to the MY-PCR reaction mix with the use of filter tips; (3) decontamination of laboratory equipment with solutions that degrade DNA; and (4) the purity of water and other reagents was regularly verified.
HPV-11 L1 protein synthesis and purification
HPV11-L1 ORF was cloned into pRSET plasmid (cat. no. V351-20; Invitrogen, Carlsbad, CA). The pRSET vectors were pUC-derived expression vectors designed for high-level protein expression and purification from cloned genes in E. coli. High levels of expression of DNA sequences cloned into the pRSET vectors were made possible by the presence of the T7 promoter. In addition, DNA inserts were positioned downstream and in frame with a sequence that encodes an N-terminal fusion peptide. This sequence includes an ATG translation initiation codon, a polyhistidine tag (HisTag) that functions as a metal binding domain in the translated protein. The HPV-11 L1 protein was produced in BL21DE3pLysS E. coli strain [One Shot® BL21(DE3)pLysS Chemically Competent E. coli, cat. no. C6060-03; Invitrogen], using isopropyl β-D-1-thiogalactopyranoside (IPTG) as expression inducer. Bacteria were cultured for 4 h after IPTG induction at 37°C, and then harvested by centrifugation. The HPV-11 L1 protein was further purified from bacterial protein extract using HisTrap Talon Metal Affinity Resin (Clontech Laboratories, Inc., Mountain View, CA) under denaturing conditions (50 mM phosphate buffer [pH 7.4], containing 8 M urea for cell resuspension and lysis; for the equilibration and washing steps 50 mM phosphate buffer containing 4 M urea was applied to the resin, and the same buffer containing 400 mM imidazol was added for protein elution). The final concentration of HPV11-L1 protein was measured using Protein Assay ESL (Roche Molecular Biochemicals, Indianapolis, IN).
Native HPV11-L1 protein was contained in bacterial extract from E. coli cells cultured at 30°C after IPTG induction in order to increase the solubility of heterologously expressed viral protein. Bacteria were lysed in the same phosphate buffer without urea, containing a mixture of protein inhibitors (inhibitor cocktail cat. no. P8465; Sigma-Aldrich, St. Louis, MO), by a repeated thawing-freezing procedure, sonication (Ultra Sonic UD-20; Techpan, Warsaw, Poland), and centrifugation (at 14,000 rpm for 30 min). The clarified bacterial extract containing a large portion of HPV11-L1 protein was used in ELISA assays after measurement of the total protein concentration (see above).
All protein electrophoreses were performed in 10% PAA gels, and proteins were detected by Coomassie blue staining. Western blots were carried out using a semi-dry blotter for protein transfer onto a PVDF membrane. For detection of HisTag HPV11-L1 protein, monoclonal anti-HisG mouse primary antibody (cat. no. R940-25; Invitrogen), and secondary goat polyclonal anti-mouse IgG antibody conjugated with horseradish peroxidase (cat. no. A4416; Sigma-Aldrich) were used. Dilutions of antibodies were prepared as described in the ELISA assays. A ready-to-use TMB solution was applied to the PVDF membrane for color development.
Detection of antibodies in antisera from patients with JORRP by ELISA assays
Microplate wells were coated with sera diluted 2× in PBS buffer (from patients with papillomas and controls), and incubated for 2 h at 37°C. All wells were washed 5 times with PBS-Teen (PBS-T) buffer, and microplate wells were filled with PBS containing 2% powdered milk to block nonspecific interactions, and they were incubated overnight at 4°C. On the second day, the wash step was repeated 5 times, and 3 μg of pure HPV11-L1 protein diluted in PBS buffer (or 50 μg of protein extract of E. coli containing HPV11-L1 protein) were added to the wells coated with human sera. The wells were incubated for 2 h at 37°C. Then the solution was removed and the wash step was repeated 5 times. Monoclonal anti-HisG mouse primary antibody (cat. no. R940-25; Invitrogen) recognizing the HisTag peptide, fused with HPV11-L1 protein was added (diluted in PBS buffer 1:5000), and they were incubated for 1 h at 37°C. The wells were washed with PBS-T buffer 5 times, and a solution of secondary goat polyclonal anti-mouse IgG antibody conjugated with horseradish peroxidase (Sigma-Aldrich) was added (diluted in PBS 1:5000), and they were incubated for 1 h at 37°C. All wells were washed with PBS-T buffer and tetramethylbenzidine (TMB; Biomeda, Foster City, CA) substrate was added, they were incubated for 20 min at room temperature, and color development was stopped by the addition of 0.5 M HCl. Optical density was read by a microplate reader (Sunrise-Basic; Tecan Group, Männedorf, Switzerland) at 490 nm. At each step of the ELISA procedure the microplate wells were filled with 200 μL of solutions/reagents.
ELISA optimization
Randomly selected and representative antisera and sera were used for ELISA test optimization, which was performed in accordance with the protocol described above as follows. Three different dilutions of antisera (undiluted, 2×, and 4× diluted) were applied, while other reagents were added to microplate wells as described above. Three different amounts of viral antigen were applied (1, 3, and 5 μg), in which case only one serum dilution was applied. Each serum sample was processed in triplicate in an intra-ELISA assay. Arithmetic mean values of optical density and standard deviations were taken to calculate the coefficient of variation (COV = SD/mean × 100). Its value was below 10% when 2× serum dilution and 3 μg of the pure HPV-11 L1 protein was applied, suggesting that the reproducibility of this assay was high. These samples were always introduced in other ELISA runs.
Cut-off values (CV) were calculated as the arithmetic mean of the absorbance values of the negative sera (MV) plus standard deviation (SD; CV = MV + SD).
The specificity of the interactions between antibodies contained in human serum, HPV-11 L1 antigen, and anti-HisTag antibody was measured by omitting the second in ELISA assays based on all tested antisera and sera. The mean optical density (OD) values in these assays were close to the mean OD value calculated for the controls obtained in the ELISA assays to which the viral antigen was added.
Such optimization was not performed with the native bacterial extract containing the HPV-11 L1 protein due to the restricted volume of sera.
Results
Total DNA isolated from oral squamous cells from patients with JORRP and controls was used as a template in PCR reactions with MY09 and MY11 primers (15). All DNA samples from patients with JORRP were HPV-positive (Fig. 1). DNA sequencing revealed that 30% of the samples were HPV6-positive, and 70% of the samples were HPV11 positive.
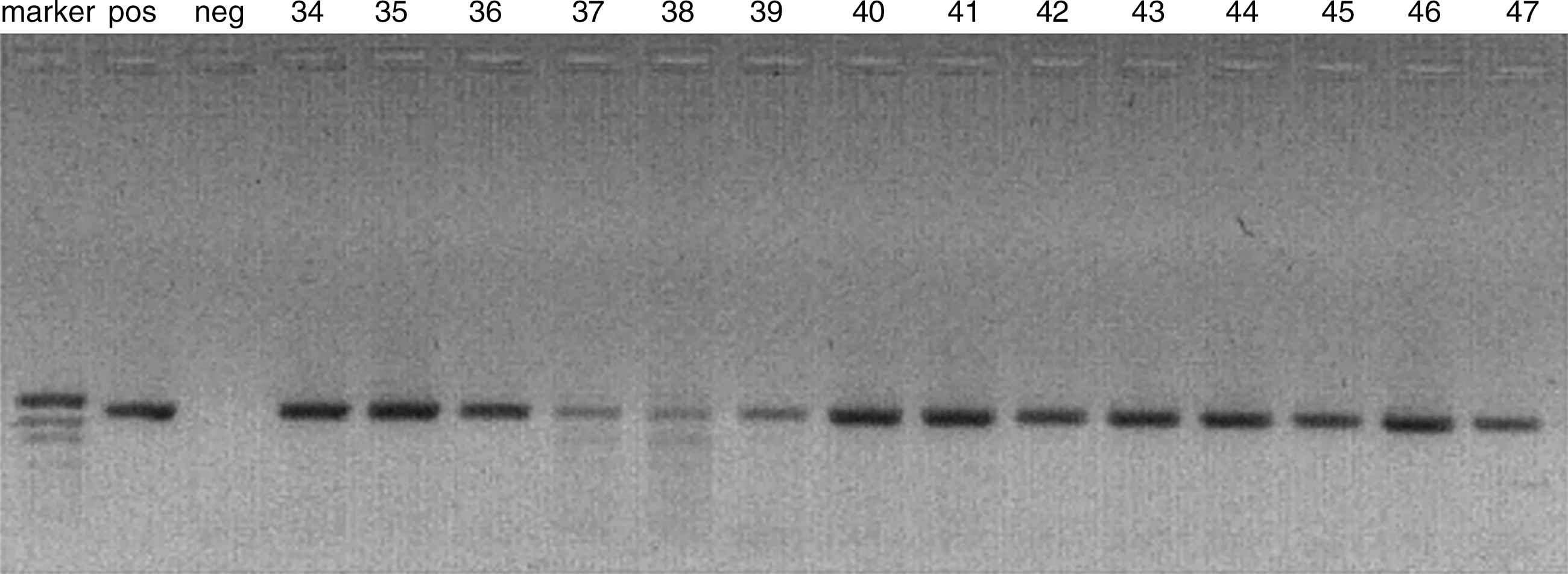
Electrophoresis in 1.5% agarose gel of MY-PCR products, synthesized with MY09 and MY11 degenerate oligonucleotides. Total DNA obtained from papillomas was used as a template (Marker, pUC19/MspI DNA molecular weight marker; Pos, positive control; Neg, negative control [DNA HPV-negative sample]; lanes 34–47, the numbers of the DNA samples from the JORRP patients; a DNA band of 450 pb is visible.
HPV11-L1 protein synthesized in E. coli was purified from other bacterial proteins under denaturing conditions, and the final concentration of the pure viral antigen was around 0.5 mg/mL. The presence of a clearly visible band of the expected molecular weight (around 58 kD: 55 kD for HPV11-L1 protein + 3 kD for HisTag) after Coomassie staining of PAA gel (Fig. 2A; lanes E1–E4), demonstrates that the purification procedure was adequate. This was further confirmed by Western blot analysis with monoclonal anti-HisG mouse primary antibody (Fig. 2B, lanes E1, E3, and E4). Prior to ELISA assays, the denatured HPV11-L1 protein solution was diluted in PBS buffer by factor of 10, as dialysis against PBS buffer resulted in HPV11-L1 protein precipitation. It is possible that a substantial fraction of the protein refolded. In our previous studies HPV11-L1 VLPs were produced and their presence was confirmed by conformational monoclonal antibodies in ELISA assays and by electron microscopy (8). In this study the research was aimed at elaborating the most simple method of viral antigen production/verification, therefore neither the presence of capsomeres or assembled VLPs were confirmed by electron microscopy or conformational antibodies.

(
In parallel, HPV11-L1 protein was produced at a lower temperature (30°C) in E. coli cells in order to increase its solubility. Native bacterial protein extract containing HPV-11 antigen was clarified as described in the materials and methods section and submitted for metal affinity chromatography in native conditions (without urea). This processing resulted in “losing” all the HPV11-L1 in the flow-through fraction, as it was not retained in the HisTrap resin, and no HPV11-L1 was detected in imidazol elution fraction (Fig. 2C). Therefore, clarified bacterial protein extract containing HPV11-L1 protein was utilized in the ELISA assays.
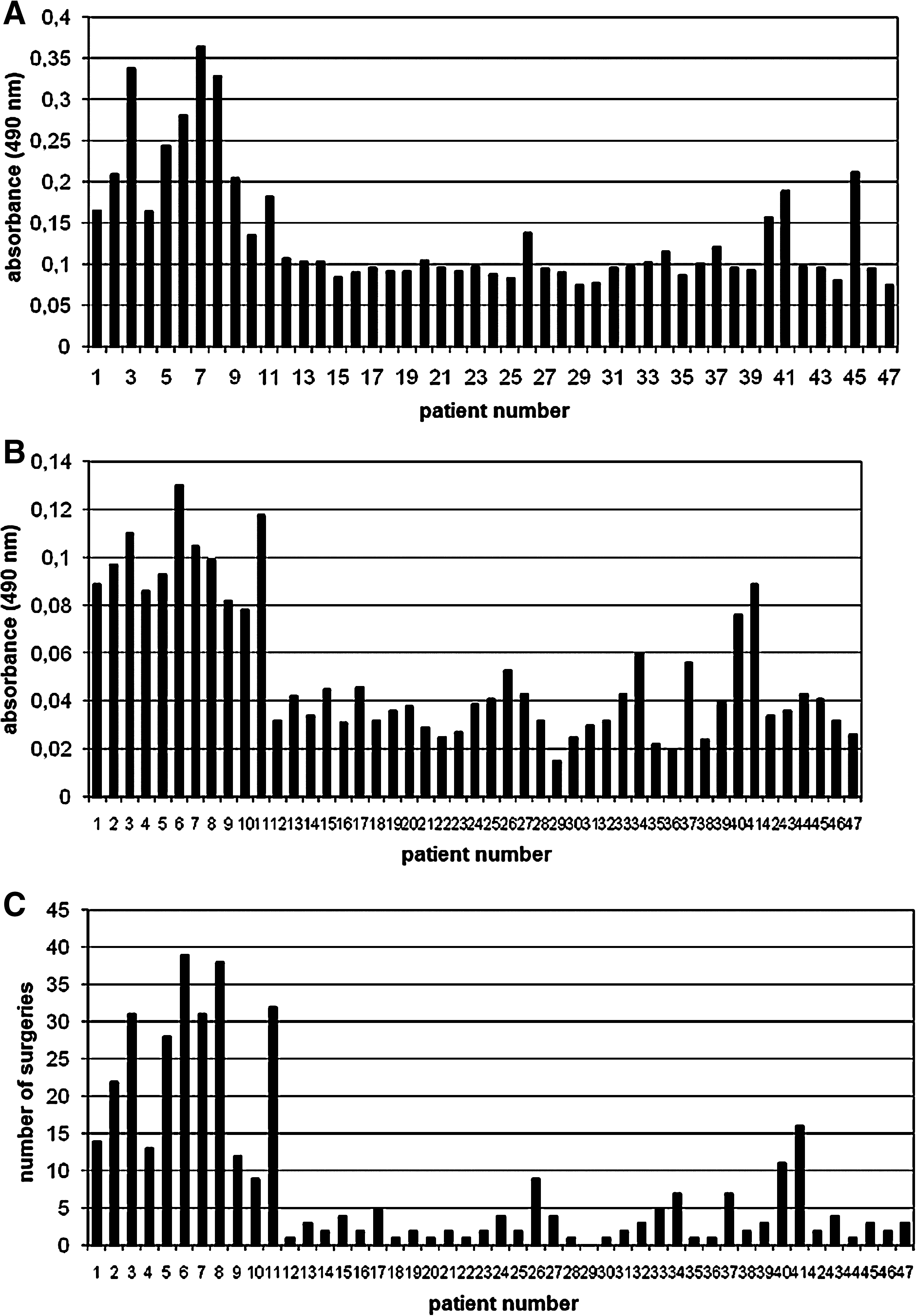
The level of anti-HPV11-L1 antibodies in antisera obtained from JORRP patients was measured in ELISA assays as previously described. Elevated OD values were seen in ELISA assays of antisera obtained from JORRP patients who had multiple surgeries to remove papillomas (the numbers of surgeries are shown in Fig. 3C). The OD values read in ELISA assays of all antisera from JORRP patients ranged from 0.075–0.364 (Fig. 3A and C), while in the control group the values ranged from 0.014–0.101, the mean value was 0.046. The cut-off value was 0.11. These results clearly show that the numbers of antibodies in the antisera directed against HPV11-L1 protein were significantly increased in patients who underwent many surgeries. For the other patients (those having only one or two surgeries) and all controls, the OD values read in the ELISA assays were similar. These results suggest that the concentration of antibodies contained in antisera directed against HPV11 virions does not differ in these groups.
(
The level of anti-HPV11-L1 antibody protein contained in antisera was also measured in the second ELISA assay using the protein contained in extracts from E. coli cells, prepared and clarified as described in the materials and methods section. This approach was undertaken in order to verify if an even simpler means of HPV11-L1 antigen preparation might be suitable for antibody detection in antisera obtained from JORRP patients. Again, elevated absorbance values were observed in ELISA assays of antisera from patients who had multiple surgeries for papillomas (Fig. 3B and C), and increased levels of antibodies approximately correlated with increasing numbers of surgeries. However, the range of OD values was smaller in ELISA assays of HPV11-L1 protein contained in native bacterial protein extract. The values obtained ranged from 0.015–0.13 for patients with JORRP, versus 0.011–0.046 for controls, for whom the mean OD value was 0.026. The cut-off value was 0.044.
The observation of correlations between the numbers of surgeries and OD values were further evaluated by employing Statistica 7.1 software (StatSoft, Inc., Tulsa, OK) to search for a mathematical description of this phenomenon. For both types of HPV11-L1 antigen used in the ELISA assays (the purified type, and that contained in clarified bacterial extract, absorbance A and absorbance B, respectively), the results were statistically significant (p < 0.001). The high values of the regression coefficients (r = 0.909 for absorbance A, and r = 0.934 for absorbance B) indicated a good match of the linear model with the empirical data (Fig. 4).

Linear regression depicting the correlation between the antibody levels detected in ELISA assays of antisera and the number of surgeries in JORRP patients. Absorbance A values were read in ELISA assays with HPV11-L1 antigen prepared under denaturing conditions and purified. Absorbance B values were read in ELISA assays with HPV11-L1 antigen prepared in native conditions, contained in clarified bacterial extract.
Discussion
The immune response against HPV is not well understood. In JORRP patients before their first surgery, as well as in asymptomatic HPV-infected people, there is only a minimal level of anti-HPV antibody in the blood. However, after multiple surgeries for papillomas in the JORRP patients, this level quickly increases. This may be due to the repeated exposure to HPV antigens in the blood when the papillomas are surgically removed. It is noteworthy that augmented antibody levels do not protect patients from re-growth of papillomas, nor do they slow their growth. It is possible that the recurrence of papillomas is strongly influenced by an altered cellular immune response. We noted that the average number of B lymphocytes was significantly increased in JORRP children, and decreased in asymptomatic carriers of HPV, compared to uninfected children. This finding, together with the substantial drop in T lymphocyte numbers in JORRP children, may be suggestive of a shift of the immune response from a cellular to a humoral one (J. Szydłowski Ph.D. dissertation, unpublished). This is an adverse phenomenon, especially in light of other findings that show little contribution by antibodies to the prevention and spread of viral infections, and this also indicates a lack of coordination of the components of the immune system. It is also consistent with other observations, namely that in RRP patients there is an alteration of both innate inflammatory signals and adaptive immune responses, both of which prevent an effective Th-1 response, along with altered expression of numerous genes that regulate cellular growth and differentiation (6).
Another question arises concerning the immune response in JORRP patients as they undergo general anesthesia for each surgical operation. It has been shown that general anesthesia has immunmodulatory effects, including changes in natural killer cell activity and T-lymphocyte subpopulations, and augmented antibody secretion in mice (9,21). Specific humoral responses can become altered [i.e., increased as described by Elena et al. (9)] after antigen administration and anesthesia; however, the latter cannot by itself influence the production of specific polyclonal antibodies.
It has also been proposed that genes coding for HLA II may play a role in JORRP development. Different HLA II alleles were identified, and a worse RRP course was seen in homozygotes bearing “defective” alleles (12).
HPV6-L1 protein is phylogenetically related to HPV11-L1 protein, and they share 92% amino acid sequence homology. The observation that anti-HPV6-L1 human antibodies recognize HPV11-L1 protein and vice versa has been noted (29). However, in our group of JORRP patients, there were no clinical cases infected with either HPV6 or HPV11 who had undergone the same number of surgeries. Therefore such precise quantitative comparison was not possible in our study. The overall HPV genotype prevalence in our study group was consistent with data published earlier, demonstrating that HPV6 and HPV11 are the dominant types in JORRP patients (12,16,20); however, in our study more cases of HPV11-related papillomas were observed (70%), whereas it was previously reported that the predominant genotype in JORRP patients was HPV6 (19). This may be due to the particular HPV epidemiology seen in our Polish population.
It is well established in the literature that HPV VLPs are more suitable for anti-HPV antibody detection, than are HPV11-L1 monomers or short immunogenic HPV epitopes (peptides) (37). We have demonstrated in our study that HPV11-L1 protein purified under denaturing conditions is a handy tool for the detection of anti-HPV11-L1 antibodies. However, a better regression coefficient was obtained in experimental approaches for HPV11-L1 protein contained in native extract. We assume that the greater OD values seen when using purified HPV11-L1 protein are due to its refolding in PBS buffer prior to immunoassays, whereas relatively small values were obtained with the native protein extracts, due to many unspecific interactions of the antibodies contained in sera from RRP patients with bacterial proteins.
In general, in ELISA assays microplates are coated with antigens (37,38), or monoclonal antibodies to capture viral antigens (23). Moreover, HPV-16 denatured antigens linked to a poly-histidine tag produced in E. coli were previously used for coating in ELISA assays of antisera from HPV-infected women (7). Coating of microplates with antisera is rarely performed, but it has been described in the literature (1,34). To challenge the above-cited studies and to further reduce financial costs, our microplates for sandwich ELISAs were coated first with human antisera, and viral antigen was added after blocking of human antibodies. Thus, the same anti-HisTag antibody could be used in Western blots for HPV11-L1 detection, and in ELISA assays as the only monoclonal antibody.
Although the coefficient of variation calculated for the ELISA assay based on antisera and purified viral antigen was satisfactory (<10%), a more thorough standardization of the method described herein would require further refinement. This was impossible for us, due to the restricted volume and relatively small numbers of antisera samples from JORRP patients.
Footnotes
Acknowledgments
Financial support for this work by the Polish Ministry of Science and Higher Education (project no. 2 P05A 049 30) was gratefully acknowledged.
Author Disclosure Statement
No competing financial interests exist.