
Abstract
Newly emerged influenza viruses have attracted extensive attention due to their high infectivity, proinflammatory actions, and potential to induce worldwide pandemics. Frequent mutations and gene reassortments between viruses complicate the development of protective vaccines and antiviral therapeutics. In contrast, targeting the host response for the development of novel cost-effective, broad-based prophylactic or therapeutic agents holds greater promise. Since inflammation often parallels the development of productive immune responses, virus-induced pulmonary inflammation and injury represents an additional challenge to the development of broad-based immunotherapeutics. Excessive inflammatory responses to respiratory viruses, also known as “cytokine storm,” are largely due to immune dysregulation that manifests as proinflammatory cytokine secretion. In addition to modulating lipid and glucose metabolism, peroxisome proliferator-activated receptors (PPAR) play important roles in antagonizing core inflammatory pathways such as NF-κB, AP1, and STAT. Their role in regulating inflammatory responses caused by pulmonary pathogens is receiving increasing attention, setting the stage for the discovery of novel applications for anti-diabetic and lipid-lowering drugs. This review focuses on the potential use of PPAR-γ agonists to downregulate the inflammatory responses to respiratory virus-related pulmonary inflammation.
Introduction
Influenza viruses are constantly evolving through a process called “antigenic drift,” which typically refers to the accumulation of mutations that alter the antigenicity of the viral glycoproteins that cover the surface of the virus particle. These minor changes are constantly accumulating during each viral replication event, due to the error-prone nature of the viral RNA polymerase, which gives rise to an ever-changing pool of virus variants. Another process called “antigenic shift,” or gene reassortment, is less common but introduces more abrupt and significant genetic changes, leading to the introduction of a new subtype of virus within a population. These evolving antigenic drifts and shifts result in new virus strains that may not be efficiently recognized by the host's immune system (3). The most recent example is the 2009 swine-origin influenza virus H1N1 strain that has rapidly spread, resulting in the WHO's declaration of a new influenza pandemic in June of 2009 (4). Contrary to predictions of a devastating influenza pandemic, the rapid spread of the virus has been associated with low mortality rates, comparable to those of currently circulating seasonal strains. However, this recent experience has confirmed that should a highly pathogenic and highly transmissible strain emerge, the lack of an adequate and timely supply of vaccines and antivirals would be a major obstacle to controlling virus spread (5).
Interventions based on vaccination, especially of high-risk groups, and early administration of antiviral drugs, are the preferred approaches for the prevention and treatment of influenza infections, respectively. Currently, it takes about 6 mo to develop a new vaccine (6). This creates a lag period between the identification of a new strain and the administration of the first vaccine doses, leaving the population exposed to the virus. The immunopathology of influenza virus is characterized by the upregulation of cytokines and other inflammatory mediators. For instance, mortality induced by the highly pathogenic H5N1 strain correlated with high levels of circulating cytokines and chemokines (7). Thus the use of agents that target the host inflammatory response rather than the virus has been proposed for the treatment of respiratory infections in general, and influenza virus infections in particular (8 –12). They offer two major advantages: first, they would be efficacious regardless of changes in viral antigenicity; and second, their production is fast and relatively inexpensive, and in the event of a highly lethal pandemic they would be readily available (13).
Targeting the Host Inflammatory Response to Treat Influenza Virus Pneumonia
The newly-emerged H1N1 influenza strain has been found to be more deadly to young individuals than the strains associated with seasonal flu (14). It has been hypothesized that the immune systems of young people (18–30 y old) is more prone to uncontrolled or hyper-immunoinflammatory responses upon infection (15). This is in line with the ability of highly pathogenic strains to induce excessive proinflammatory cytokine secretion. Influenza A (H5N1) and the reconstructed influenza A/1918 (H1N1) trigger an exacerbated inflammatory response in humans that died from the infection, and in mice inoculated experimentally (7,16). Pathological examination of lung tissue from fatal cases of the 2009 swine-origin H1N1 strain support these findings (17). If this hypothesis is correct, then the therapeutic interventions to minimize influenza-related pathology and mortality require suppression of the excessive inflammatory response, without hindering the induction of protective influenza virus–specific immunity. However, knockout mice lacking key cytokines or chemokines that participate in this response (i.e., MIP1-α, IL-6, and TNF-α), or their receptors (i.e., IL-1βR), were either unaffected or even more susceptible to the disease. It has been argued that the existence of functional redundancy between these inflammatory mediators and their signaling pathways could explain this apparent contradiction (18,19).
The use of the class of cholesterol-lowering drugs known as statins as an anti-inflammatory intervention for influenza virus–related lung pathology was first proposed in 2005 (9). However, published data on the putative benefits of statins for the treatment of viral pneumonia originated from observational studies. Specifically, patients treated with statins were less likely to be hospitalized or to die from influenza viral infections (20,21). In contrast, other groups have shown only marginal benefits associated with statin therapy (22). In this context, data from human clinical trials and mechanistic studies in animals are required to investigate the therapeutic and prophylactic efficacy of statins against influenza.
Another class of drugs used to treat metabolic disorders is the nuclear receptor peroxisome proliferator-activated receptor (PPAR) agonists. Because PPAR activation antagonizes inflammatory pathways, PPAR agonists could be used to downregulate the host inflammatory response to influenza virus. Budd et al. reported in 2007 that the PPAR-γ agonist gemfibrozil, a drug prescribed to lower plasma lipids and cholesterol, increased survivability of mice infected with influenza virus A/Japan/H2N2 from 11% to 50% (23). More importantly, gemfibrozil was able to ameliorate established viral disease, since the treatment was given 4 days after infection, when the mice already exhibited clinical signs. PPAR-γ is another isoform that suppresses inflammation. Recently, Aldridge et al. demonstrated that the prophylactic administration of pioglitazone, a synthetic PPAR-γ ligand, reduced mortality of mice infected with influenza A/PR8/H1N1 virus from 92% to 50% (24). Other PPAR-γ agonists have shown potential beneficial effects for the treatment of RSV infections. For instance, 15d-PGJ2, ciglitazone, and troglitazone decreased the production of the proinflammatory cytokines IL-1α and TNF-α, and the chemokines CXCL8 and CCL5, as well as GM-CSF, IL-6, and intercellular adhesion molecule-1 of RSV-infected epithelial cells (25,26). Moreover, PPAR-γ agonists inhibited viral G, F, and N protein expression and RSV replication in lung epithelial cells in vitro (27). It was also shown that late treatment with antivirals (i.e., 48 h post-infection) reduced mortality in mice inoculated with influenza A/H5N1 virus only when co-administered with the immunosuppressive drugs celecoxib and mesalamine (28). The improvement in survivability was not associated with decreased viral loads, but with inhibition of cyclooxygenase-2. One of the immunomodulatory drugs used in this study, mesalamine, is also a PPAR-γ agonist (11,29). Altogether, these data suggest that the suppression of inflammatory responses by PPAR-γ agonists could represent a viable therapeutic or prophylactic strategy against flu and other respiratory infectious diseases.
PPAR-γ Overview
Peroxisome proliferator-activated receptors (PPARs) belong to the superfamily of nuclear hormone receptors with 48 members identified in the human genome. There are three known PPAR isoforms: α, β or δ, and γ, which differ in their tissue distribution and functional activity (30), and all three are expressed in the lungs (31). PPAR-γ is an endogenously controlled molecular switch that regulates inflammation, immunity, and metabolism (32,33). This particular isoform was initially studied as a key regulator of lipid and glucose homeostasis by promoting adipocyte differentiation (34,35), and was identified as the molecular target for the insulin sensitizing thiazolidinediones (TZD) (36). However, in the last 10 years, research has also focused on the anti-inflammatory actions of PPAR-γ. Administration of exogeneous ligands of PPAR-γ, including TZD, has proven to be beneficial in several animal models of autoimmune and inflammatory diseases (37 –40), and in human clinical trials with ulcerative colitis patients (41).
PPAR-γ activation and expression is also controlled by a diverse set of natural and synthetic molecules, including nutrients, non-nutrient endogenous ligands [i.e., prostaglandins or hydroxy-containing PUFA such as 12/15-hydroxyeicosatetraenoic (12/15-HETE), 13-(S)-hydroxyoctadecadienoic (13-HODE), and conjugated linoleic acid (CLA)], and drugs [e.g., TZD and 5-aminosalicylic acid] (42 –44). In addition, certain cytokines like IL-4 upregulate PPAR-γ expression (45), whereas others, like TNF-α, suppress it (46). This regulatory network provides a functional link between PPAR-γ activity at the cell or tissue scales, and the state of immune activation or inflammation. PPAR-γ exerts its function mainly via a ligand-dependent activation. Ligand binding induces PPAR and retinoid X receptor (RXR), forming a heterodimer (47). This activated PPAR-RXR heterodimer translocates into the nucleus, where it binds peroxisome-proliferator response elements (PPRE) located in the regulatory area of PPAR target genes. In this way ligand-activated PPAR-γ regulates the transcription of a set of genes involved mainly in glucose homeostasis and lipid uptake and metabolism (35,48). In addition, activated PPAR-γ can suppress the expression of specific subsets of proinflammatory proteins by interfering with the transcription factors that regulate their expression, such as NF-κB or MAPK (49,50).
In contrast to the induction of target genes, PPAR-γ transrepression activity does not require direct binding of the activated form to the DNA in promoter regions of the affected genes. A well-characterized example is the suppression of LPS-induced iNOS by rosiglitazone-activated PPAR-γ in mouse macrophages. The ligand-dependent sumoylation of PPAR-γ targets it to nuclear receptor co-repressor complexes on inflammatory gene promoters, which serves to stabilize and prevent signal-dependent removal of co-repressor complexes (51). Sumoylation of PPAR-γ induced by apoptotic cells has also been described in macrophages, which results in downregulation of TNF-α and IL-6 expression (52). Graphical illustrations of the sumoylation of PPAR-γ by rosiglitazone have been published by Glass et al. (51). In addition to ligand-dependent activation, PPAR-γ activity can also be modulated by phosphorylation, which may change the affinity of PPAR-γ for its ligands (53).
PPAR-γ Expression and Function in the Lungs
PPAR-γ is expressed in healthy lungs in the airway epithelium, bronchial submucosa, vascular smooth muscle cells, and endothelial cells (54). In addition, immune cells, including T cells, B cells, dendritic cells (DCs), neutrophils, eosinophils, and macrophages express PPAR-γ during differentiation or following activation. As a general rule, PPAR-γ expression increases as cells transition from an immature to a differentiated state. For instance, PPAR-γ is expressed upon differentiation of monocytes into macrophages (55,56). In addition, the healthy alveolar macrophage expresses more PPAR-γ protein constitutively than other macrophage subsets (57). At the organ level, lung PPAR-γ expression is upregulated following allergen sensitization of mice, and in the airways of asthmatics (58). Moreover, treatment with rosiglitazone caused a modest 15% improvement in the allergen challenge model in asthmatic patients (59). In contrast, endotoxin-induced lung injury was associated with lower PPAR-γ expression (54). As for other inflammatory conditions, several chronic lung pathologies including asthma, chronic obstructive pulmonary disease (COPD), and lung fibrosis (60), are ameliorated by exogenous administration of PPAR agonists. For instance, treatment of human lung fibroblasts with PPAR-γ agonists in vitro blocked their differentiation into myofibroblasts induced by exogenous TGF-β, and consequently diminished the collagen deposition that leads to fibrosis (61). Also, the administration of the TZD pioglitazone via aerosol lowered antigen-induced hyperresponsiveness, BAL eosinophilia, type II cytokines, and IgE levels (61 –63). Thus, PPAR-γ is now being investigated as a potential target in a variety of lung-related chronic inflammatory diseases. However, the role of PPAR-γ and its agonists in regulating lung inflammation during respiratory microbial infections remains largely unknown.
Immune Cells Involved in Influenza-Related Pulmonary Pathogenesis and the Potential for Regulation by PPAR-γ
The host response induced by influenza virus is characterized by the activation of epithelial cells and alveolar macrophages, followed by the recruitment of inflammatory leukocytes into the pulmonary parenchyma (1,64). This response is associated with lung pathology, and in highly pathogenic influenza infections, with increased mortality (16 –18). In parallel, the innate immune response is necessary for virus clearance and for the induction of productive CD8+ T-cell responses. Thus the ideal immunomodulator should lower inflammation-induced pathology while still maintaining a microenvironment that is conducive for establishment of the virus-specific acquired immunity necessary to successfully overcome the infection. Aldridge et al. showed that while blockade of leukocyte recruitment via deletion of CCR2 leads to increased mortality, moderate suppression of MCP-1 (i.e., the ligand of CCR2) by exogenous activation of PPAR-γ was beneficial. At the cellular level, the mechanism of protection depended on the decreased presence of a subset of DCs that secrete proinflammatory cytokines (tipDC) (65), and are also needed to induce/maintain virus-specific CD8 T cells in the lung (24). Thus the use of PPAR agonists or other immune modulators could dampen the negative consequences of the aggressive host response, without having a blunt effect in the induction of protective immunity to the virus. The following section reviews the cell types that contribute to the pulmonary inflammatory response and their regulation by PPAR-γ and its agonists.
Respiratory epithelial cells
Respiratory epithelial cells, including alveolar and bronchial epithelial cells, are the primary cellular targets and first line of innate immune defense to influenza virus. Together with alveolar macrophages and DCs, epithelial cells detect influenza viruses via PRRs (66). Infected epithelial cells produce several proinflammatory factors, which can modulate the immune response as well as lead to tissue damage. Guillot et al. showed that influenza A virus activates toll-like receptor 3 (TLR-3) signaling, leading to the secretion of proinflammatory mediators such as IL-8, IL-6, regulated on activation, normal T-cell expressed and secreted (RANTES), and IFN-β (67). Various strains of influenza viruses elicit different profiles of cytokines and chemokines. For instance, compared to influenza A (H1N1), the H5N1 virus tends to elicit excessive production of IP-10, IFN-β, RANTES, and IL-6 in epithelial cells (68), whereas H3N2 strains exhibit milder inflammatory responses. The secretion of immune modulators and antigen presentation makes respiratory epithelial cells extremely important in immune defense following influenza virus infection.
The death rate of alveolar epithelial cells can be as much as 15% 24 h after infection (69). Inhibition of host mRNA translation in infected epithelial cells is one of the major causes of epithelial cell death. PB1-F2 protein of influenza virus is demonstrated to induce cell death, including apoptosis of alveolar epithelial cells, via a mitochondria-related mechanism (70). TNF-related apoptosis-inducing ligand (TRAIL) expressed by macrophages also contributes to alveolar epithelial cell apoptosis (71). Various cytokines also induce apoptosis in alveolar epithelial cells. Production of proinflammatory cytokines is reduced during epithelial cell apoptosis (72). Therefore, respiratory epithelial cell apoptosis is important to eliminate infected and damaged cells, as well as to control inflammation and therefore maintain host responses, while minimizing inflammation-induced tissue damage.
Airway epithelial cells are the first barrier of the respiratory system and the first to respond to infection. PPAR-γ expression in airway epithelial cells is constitutively high and will inhibit the expression of inflammatory factors such as inducible nitric oxide synthase (iNOS) and IL-8. Agonists of PPAR-γ, such as the prostaglandin D2 metabolite 15-deoxy-Δ12,PGJ2 and ciglitazone, enhance this inhibition in a dose-dependent manner, as do other PPAR γ ligands (73). Therefore, the use of PPAR-γ agonists to activate PPAR-γ as an inflammatory modulator is prominent in the literature. However, the possible application of PPAR-γ therapy to suppress influenza-related inflammation is a novel and a promising therapy in the face of current and emerging pandemics.
Respiratory macrophages
Monocytes/macrophages play a central role in immune regulation in response to pathogens, as well as maintaining pulmonary homeostasis. The alveolar macrophage is the most important subset of respiratory macrophages, accounting for the majority of immune cells in alveolar spaces. Infiltration of lung tissue by newly differentiated macrophages is markedly increased in influenza virus–infected humans and mice, which suggests a possible role of this immune cell subset in inflammation and lung injury, as well as in eliciting an adaptive immune response (74).
Although the alveolar macrophage contributes to lung homeostatic regulation in part by suppressing the induction of immune responses in healthy individuals, following influenza virus infection they acquire a proinflammatory phenotype. They respond to virus infection by producing proinflammatory cytokines such as TNF-α, IFN-β, IFN-λ, IL-6, IL-1β, IP-10, CCL5/RANTES, CCL2/MCP-1, CCL3/MIP-1α, and CCL4/MIP1-β (74 –76). IFN regulatory factor 3 (IRF3) and p38 kinase work separately to mediate H5N1-induced TNF-α, IFN-β, IFN-λ, and MCP-1 (77).
During the steady state, resident alveolar macrophages are maintained at stable levels and are mainly renewed by precursor cell proliferation. They function as phagocytes, but do not migrate to regional lymph nodes, which constitute the primary place for initiating adaptive immunity and the inductive sites of the respiratory mucosa (78). During viral-induced inflammation, recruited macrophages originate from peripheral blood monocytes, move across the alveolar epithelium, and infiltrate the alveolar spaces. Herold et al. demonstrated that it is the alveolar epithelial cell rather than resident alveolar macrophages that release monocyte chemoattractants such as CCL2/MCP-1 and CCL5 (79).
Alveolar macrophages regulate the pulmonary immune response (80), and can suppress APC function of pulmonary DCs (81). The mechanism behind the suppression of alveolar macrophages to reduce innate and adaptive immunity is not quite clear. Morris et al. found that it may be related to αvβ6 integrin expression in alveolar epithelial cells, a process regulated by TGF-β (82). In addition, high levels of PPAR-γ expression by alveolar macrophages had a suppressive role in inflammatory cytokine secretion (83), as well as AP-1 and NF-κB activation (84), which suggests a possible PPAR-γ-related mechanism.
Alveolar macrophages have also been reported to exert a modulatory role on DC migration (85). As noted previously, infiltrating exudate macrophages via release of TRAIL can also potentiate respiratory epithelial cell death, leading to exacerbated lung injury and increased mortality (71). Lin et al. reported increased numbers of CCR2+ monocyte-derived DCs and exudate macrophages in the lungs of influenza virus–infected mice, which produce large amounts of cytokines and could be the major cell types contributing to influenza virus–induced pulmonary inflammation and pathology. Of note, CCR2-knockout mice have decreased macrophage infiltration and pulmonary injury (86,87).
Alveolar macrophages highly express PPAR-γ, which is even further upregulated by IL-4 (88) and GM-CSF (89). Treatment with a PPAR-γ agonist in vitro showed upregulated expression of PPAR-γ, and suppressed production of proinflammatory cytokines such as TNF-α and IL-12 (83), as well as AP-1, and NF-κB (84). In addition, PPAR-γ knockout in macrophages leads to a significantly increased expression of IFN-γ, iNOS, IL-12, MIP-1α, and IP-10, which are Th-1 immune mediators (90). PPAR-γ has also been found to regulate macrophage colony-stimulating factor (M-CSF), which is a mediator of monocyte differentiation, activation, and survival, via transrepression of NF-κB (91). These observations suggest that PPAR-γ activation in alveolar macrophages elicits an anti-inflammatory effect, though the mechanism is not yet fully understood. Of note, MCP-1 production is reduced by administration of PPAR-γ ligands (92). Thus PPAR-γ may exert its anti-inflammatory function partially by indirectly regulating exudate macrophage infiltration via its ability to limit MCP-1 production in alveolar macrophages.
Dendritic cells
As professional antigen-presenting cells (APCs), DCs are found in relatively small amounts at mucosal surfaces like skin or the inner layer of the lungs and intestinal lamina propria. DCs directly interact with T and B lymphocytes at inductive sites (i.e., mediastinal lymph nodes) to initiate adaptive immune responses and serve as sentinels of the immune system. In this regard, a newly characterized subset of DCs that produce TNF-α and iNOS, known as tipDCs (65), has been shown to contribute to both influenza virus–associated pulmonary inflammation, as well as to the induction of protective immune responses (24). tipDCs accumulated in higher numbers in the lungs of mice infected with highly pathogenic strains than in mice inoculated with strains that induce milder disease (24). Recruitment of monocytes and differentiation into tipDCs depends on CCR2 signaling (93). Very few tipDCs were found in lungs of CCR2–/– mice; however, this was paralleled by poor induction of influenza virus–specific CD8+ T cells and inefficient virus neutralization and clearance. The synthetic PPAR-γ agonist pioglitazone suppressed without fully inhibiting tipDC accumulation, thereby moderating the morbidity and mortality of the influenza virus infection, while maintaining the ability to mount adaptive immune responses (24). While the expression of iNOS by murine macrophages is well documented, the generation of NO by human macrophages has been difficult to demonstrate, thereby limiting the translational value of tipDCs.
Aldridge et al. found that the main difference between pioglitazone and vehicle-treated animals was the lower levels of MCP-2, which correlated with decreased presence of tipDCs. However, the lower numbers of these proinflammatory cells was not associated with decreased overall iNOS expression in the lung (24). Moreover, although tipDCs are important for inducing and/or maintaining CD8+ T cells in the lung parenchyma, there were no differences in viral loads, indicating that the enhanced CD8+ T-cell response did not correlate with productive antiviral immunity. In addition to regulating the recruitment of DCs, PPAR-γ ligands can also modulate the induction of adaptive immune responses by inhibiting DC maturation, the expression of co-stimulatory molecules, and the secretion of IL-12 (94,95). During in vitro differentiation of human monocyte-derived DCs (moDCs), PPAR-γ is expressed only briefly in the initial phases of the differentiation process. This window of PPAR-γ activity coincides with the upregulation of genes involved in lipid uptake and metabolism (56). Moreover, DC differentiation in the presence of the PPAR-γ agonist rosiglitazone suppressed the expression of CD1a, CD1b, CD1c, and CD1e (i.e., group I lipid-presenting molecules), whereas CD1d was significantly upregulated. CD1d, which is normally present in monocytes but not in DCs, presents α-galactosylceramide (α-GalCer) to suppressor natural killer T (NKT) cells, facilitating their expansion (96). Co-administration of α-GalCer as an adjuvant with inactivated influenza vaccine diminished the initial CD8+ T-cell response, but in the long term enhanced the generation of memory CTL and protective immunity (97).
T lymphocytes
CD4+ T cells can promote the proliferation and differentiation of T cells, B cells, and other immune cells by secreting IL-2. CD4+ T cells are functionally heterogeneous, and include Th-1, Th-2, and Th17, and naturally occurring as well as induced regulatory T cells (Treg). PPAR-γ activation has been shown to downregulate the production of IFN-γ, IL-17, and IL-2 by CD4+ T cells, thereby resulting in suppressed proliferation and Th-1 or Th-17 differentiation (98,99). In addition, the expression of functional PPAR-γ in naturally-occurring Treg is required for the prevention of effector CD4+ T-cell-induced chronic colitis (99), and graft-versus-host disease (39). In the model of virus-induced respiratory disease, IFN-γ may contribute to both facilitating viral clearance, as well as augmenting the lung inflammatory response. Therefore, activation of PPAR-γ would be beneficial in ameliorating CD4+ T-cell-driven pulmonary inflammation, but could possibly impair Th1-associated antiviral immunity. Therefore, treatment with PPAR-γ agonists should be considered as a prophylactic therapeutic for use against those viral strains that cause respiratory disease through pulmonary inflammation.
CD8+ T cells include suppressor or regulatory T lymphocyte (Ts) and cytolytic T lymphocyte (Tc) cells. There are recent reports of Tc17 CD8+ T-cell effectors that contribute to protective immunity against influenza (100). CD8+ T cells play an important role in viral clearance and recovery from viral infection. CD8+ T-cell-deficient mice show impaired viral clearance and therefore higher mortality after influenza virus infection (101). Transfer of CD8+ T cells to B cell-deficient mice can facilitate the clearance of influenza virus from the lungs (102). IFN-γ produced by CD8+ T cells is found to play a critical role in the CD8+ T-cell response, since transfer of CD8+ T cells from IFN-γ-deficient mice does not result in immune protection (103). During influenza virus infection, immediately after the induction of innate immune responses, activated antigen-presenting DCs migrate to the mediastinal lymph nodes and activate antigen-specific naïve CD4+ and CD8+ T cells (104). Migration of the virus-specific CD4+ and CD8+ T cells, which can express IL-2, IL-4, IL-10, and IFN-γ (105), back to the lung is largely dependent on the chemokines expressed in the lung during the infection (106,107). In line with the concept that regulatory mechanisms parallel proinflammatory responses, Sun et al. found that during acute influenza virus infection, CD4+ and CD8+ effector T cells produce high levels of IL-10, an important anti-inflammatory cytokine that is reported to be produced by several immune cell types in response to inflammatory stimuli (108,109). Blockade of IL-10 leads to more severe pulmonary inflammation during influenza virus infection. In addition, an increasing number of IL-10-producing CD8+ T cells at the infection site indicates the possible modulatory role of CD8+ T cells during influenza virus infection. Altogether, virus-specific effector T cells play a significant role in controlling influenza virus infection by inducing infected cell lysis and the generation of inflammatory mediators, as well as contributing to pulmonary homeostasis through the induction of regulatory responses. Even though no information is available on the direct regulation of CD8+ T-cell responses by PPAR-γ compared to PPAR-γ-mediated regulation of CD4+ T-cell responses, a recent report suggests that PPAR-γ activation of DCs results indirectly in impaired activation of antigen-specific CD8+ T cells by inducing the upregulation of the co-inhibitory molecule B7H1 (110). Further studies are needed to examine the role of PPAR- γ in CD8+ T cells during respiratory viral infection.
Neutrophils
Neutrophils constitute a large proportion of the inflammatory leukocytes present in the lung following influenza virus infection. Although their role in influenza virus clearance is not yet well defined, it has been suggested that they may hinder influenza virus replication (111). Also, influenza A virus infection can increase apoptosis of neutrophils, which might contribute to the inflammatory response (112). Wareing et al. also showed that inhibition of neutrophil recruitment did not have a significant effect on viral clearance (113). However, in a different study, neutrophil depletion aggravated lung pathology, leading to fatal pneumonia and death in mice infected with a low dose of a sublethal H3N2 influenza virus (114). In the absence of neutrophils, the virus spread to extrapulmonary locations. Thus, similarly to the inflammatory tipDCs, modulation of neutrophil recruitment and function by PPAR-γ could lead to diminished pathology without having adverse effects on the control of virus spread and persistence. Human neutrophils express moderate levels of PPAR-γ that can be increased by TNF-α and IL-4 (115). While the upregulation of mRNA and protein by IL-4 is common to other cell types, the effect of TNF-α seems to be specific for neutrophils, as others have reported an inhibitory effect on the expression and/or activation of PPAR-γ by this proinflammatory cytokine in other immune cells (116). In addition, PPAR-γ-agonist treatment was shown to inhibit neutrophil chemotaxis (115). Lipoxin A4 is an eicosanoid that plays a key role in the resolution of neutrophilic inflammation. However, decreased PPAR-γ activity in neonatal neutrophils correlated with reduced responsiveness to lipoxin A4 (117). Ciglitazone, another PPAR-γ agonist, was found to significantly impair neutrophil infiltration into the lung via a NF-κB-related mechanism (118). Although these findings are not directly related to influenza-associated inflammation, they do suggest that PPAR-γ also modulates the anti-inflammatory responses in neutrophils, providing an additional regulatory target for PPAR-γ-based immunotherapeutics.
Conclusion
Traditional antiviral therapeutics may offer limited efficacy due to the rapid evolution of emerging influenza virus strains, and the emergence of strains resistant to antiviral treatment. Since excessive inflammation has been linked to influenza virus-related mortality, immune modulation may be required in addition to virus clearance to limit or prevent severe disease. Exogenous administration of PPAR-γ ligands can broadly target the majority of cells involved in virus-induced pulmonary pathology (Fig. 1), and it represents an effective means of immune modulation. Its transrepression of inflammatory genes significantly modulates the immunological networks at the respiratory mucosa, and therefore exerts prophylactic and therapeutic effects during respiratory virus infection. The specific mechanism by which PPAR-γ agonism protects from influenza-induced lung pathology remains unknown and additional studies are required. For instance, although pioglitazone diminished the recruitment of TNF-α- and iNOS-producing DCs, the production of NO in human myeloid-derived cells has been difficult to demonstrate, with the exception of Mycobacterium tuberculosis infection (119,120). The lack of parallelism between mice and humans may call into question the translational value of these findings. However, PPAR-γ is expressed in several compartments and cell types of the lung, and it is likely that epithelial cells are also targeted by PPAR-γ-based interventions. In non-human primates infected with avian-origin H5N1 influenza A, induced virus replication was mainly localized to type II pneumocytes, and resulted in strong induction of IL-6 and TNF-α (121). Of note, both of these proinflammatory cytokines are downregulated following PPAR-γ activation. Thus it is likely that the mechanism of action of PPAR-γ agonists involves both the epithelial and immune compartments. In addition, many agonists activate PPAR-γ-independent pathways, thus the use of genetically modified, conditional PPAR-γ-deficient mice in immune, epithelial, or endothelial cells will aid in elucidating the cell specificity and mechanism of action of such putative therapy.
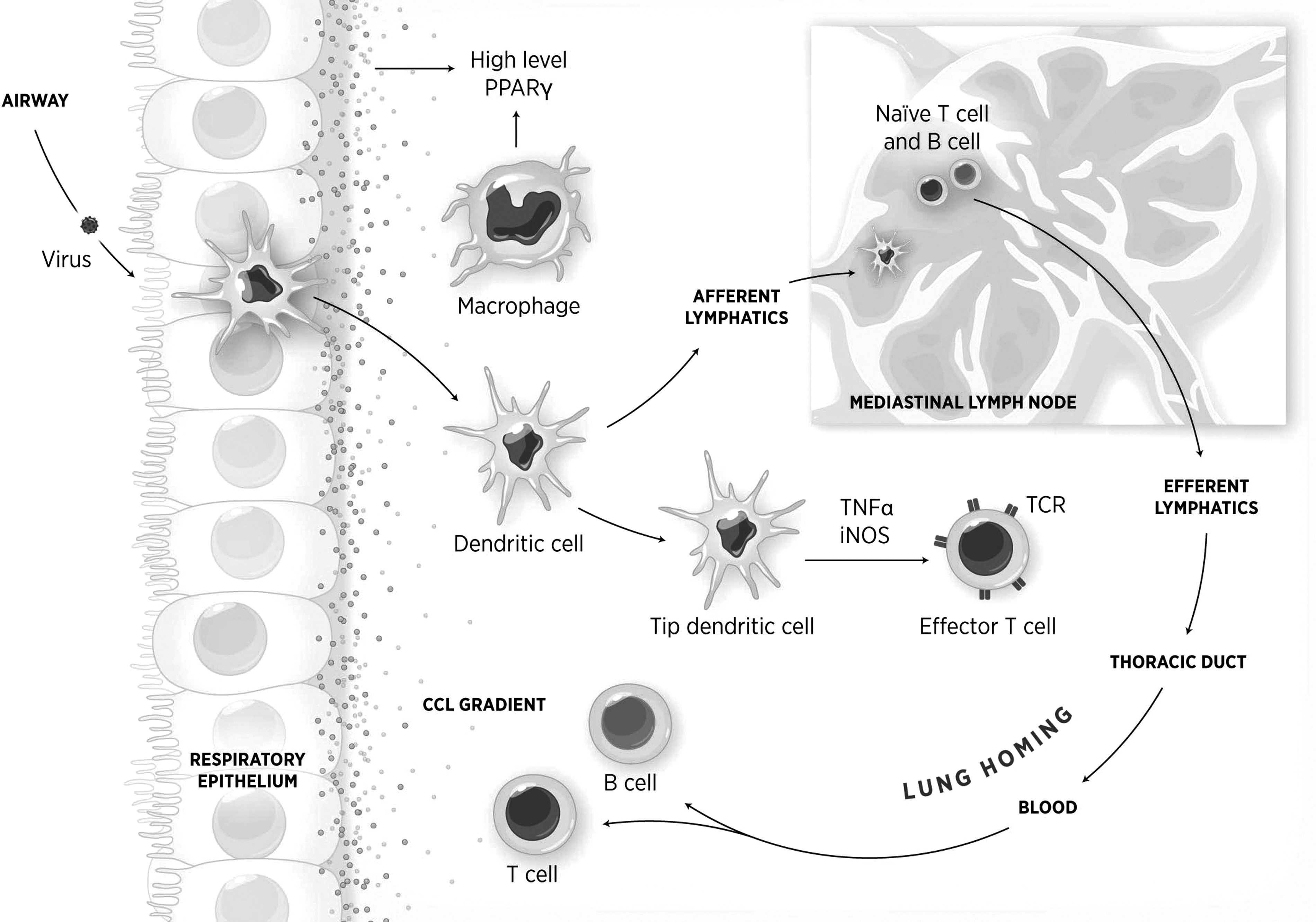
Epithelial cells are the first line of defense against respiratory viruses. They detect virus via pattern recognition receptors (PRRs) and produce proinflammatory factors. Following infection, bone marrow–derived macrophages infiltrate the lung area. Recruited and resident macrophages can regulate pulmonary inflammation. Although present in relatively small amounts in the respiratory mucosa, dendritic cells (DCs) migrate to the mediastinal lymph nodes and present antigen to naive T and B cells, which in turn will migrate to the lungs as effector cells following a chemokine (CCL) gradient. tipDCs migrate into the lung during influenza virus infection and mediate inflammatory responses. Epithelial cells, DCs, macrophages, and T cells express peroxisome proliferator-activated receptor-γ, and its activation results in amelioration of virus-induced pulmonary inflammation.
Footnotes
Acknowledgments
We would like to thank Ms. Jennifer Wang for her assistance in the preparation of the figure.
Author Disclosure Statement
No competing financial interests exist.