
Abstract
Disease activities of hepatitis B are affected by the status of hepatitis B e antigen (HBeAg). The function of the hepatitis B virus (HBV) precore or HBeAg is unknown. We assumed that HBeAg blocks aberrant immune responses, although HBeAg is not required for viral assembly, infection, or replication. We examined the interaction of HBeAg and the immune system, including cytokine production. The inflammatory cytokine TNF, IL-6, IL-8, IL-12A, IFN-α1, and IFN-β mRNA were downregulated in HBeAg-positive HepG2, which stably expresses HBeAg, compared to HBeAg-negative HepG2 cells. The results of real-time RT-PCR-based cytokine-related gene arrays showed the downregulation of cytokine and IFN production. We also observed inhibition of the activation of NF-κB- and IFN-β-promoter in HBeAg-positive HepG2, as well as inhibition of IFN and IL-6 production in HBeAg-positive HepG2 cell culture fluids. HBeAg might modify disease progression by inhibiting inflammatory cytokine and IFN gene expression, while simultaneously suppressing NF-κB-signaling- and IFN-β-promoter activation.
Introduction
The HBV genome consists of four open reading frames coding for the surface, core, polymerase, and X proteins. Viral DNA, upon entry into cells during productive infection, undergoes a repair process and forms covalently closed circular DNA. Transcription of this DNA produces longer (precore) and shorter (pregenomic) 3.5-kb RNAs. The pregenomic RNA is packaged into nucleocapsides along with the viral polymerase, and serves as the template for viral genome replication. Precore and pregenomic RNAs encode core, polymerase (by pregenomic RNA), and hepatitis B e antigen (HBeAg) (by precore RNA) (47).
Disease severity of hepatitis B is affected by the status of HBeAg. The presence of HBeAg in serum is also known to be a marker of a high degree of viral infectivity. Although there are diverse opinions, fulminant hepatitis may occur in persons who are negative for HBeAg in highly endemic areas (29). Infants born to HBeAg-positive mothers tend to be HBsAg-positive more than those born to HBeAg-negative mothers (44). HBeAg-positive asymptomatic carriers (ASCs) have higher viral load, but most do not display any liver dysfunction (10). These clinical cases can be assumed to have immune tolerance for HBeAg.
The core gene of 183 codons (at least for genotypes B and C) is preceded by an in-frame pre-ATG codon that extends the protein by 29 hydrophobic amino acids (Fig. 1A). Proteins like this are translated from a 3.5-kb precore RNA and converted to HBeAg by two proteolytic cleavage events in the secretory pathway (12,26,38). First, the N-terminal 19 residues encoded by the precore region serve as the signal peptide for translocation of the precore/core protein into the endoplasmic reticulum lumen, where the peptide is clipped away by a signal peptidase. Next, 30 residues are removed from the C terminus in a post-endoplasmic reticulum compartment to generate mature HBeAg of ∼17 kDa (12). A single point mutation has been reported to produce a stop codon in the precore region of HBV DNA and prevent the formation of the precore protein required to make HBeAg (7). HBeAg is thought to involve immune tolerance via an unknown mechanism, although it is not required for viral assembly, infection, or replication (3,42). Visvanathan et al. (43) reported that the expression of TLR2 on hepatocytes, Kupffer cells, and peripheral monocytes, was significantly reduced in HBeAg-positive chronic hepatitis B patients. Although the precise function of HBV precore or HBeAg is unknown, it is possible that HBeAg suppresses the TLR pathways, thereby allowing HBV to establish persistent infection in the host (43).

(
Toll-like receptors (TLRs) play important roles in the innate immune response and are thought to have therapeutic potential for infectious diseases and cancers (18). Some of them are expressed on many different cells, including hepatocytes (32,36). Preiss et al. (32) demonstrated mRNA transcription for most TLRs, with the exception of TLR8. TLR5 mRNA was not detectable in HepG2 cells. Hepatocytes may themselves play an active role in innate immune responses to viruses such as HBV (32). Once these pattern recognition receptors (PRRs) have identified the pathogen-associated molecular patterns (PAMPs), the effector cells function and respond immediately. Ligand recognition by TLRs leads to the recruitment of various TIR domain-containing adaptors, such as myeloid differentiation primary response gene (88) (MyD88), toll-interleukin 1 receptor domain containing adaptor protein (TIRAP), TIR domain-containing adapter inducing interferon-β (TRIF), and TRIF-related adapter molecule (TRAM), which in turn triggers the cascade of the signaling pathway, and ultimately the activation of transcription factors such as nuclear factor-κB (NF-κB) and interferon regulatory factors (IRFs), leading to the expression of various cytokines (e.g., tumor necrosis factor [TNF], interleukin-6 [IL-6], IL-8, interferon-α1 [IFN-α1], and IFN-β). Hepatic cytokines also play an important role in the progression of hepatitis B-associated liver diseases. A number of viruses have been shown to encode proteins that have the potential to inhibit antiviral activity of the innate and adaptive immune responses. Inflammatory cytokines contributing to viral clearance in HBV infection may have therapeutic value (20). In the present study, we assumed that HBeAg blocks aberrant immune responses, and we examined the role of HBeAg protein in cytokine production to test the interaction between HBeAg and the immune system in human hepatocytes. Our results demonstrated that cytokine production is inhibited by HBeAg, and that it also enhances IFN-sensitive hepatitis C virus (HCV) replication.
Materials and Methods
Plasmids
pNF-κB-luc, which expresses luciferase upon promoter activation by NF-κB, was purchased from Stratagene (La Jolla, CA). This vector has five repeats of the binding site for NF-κB (TGGGGACTTTCCGC). pIFN-β-luc, which expresses luciferase under the control of an IFN-β-dependent promoter, was kindly provided by Dr. N. Kato (Institute of Medical Science, University of Tokyo, Japan). To construct plasmids including HBV precore and core regions, HBV DNA was used from the serum of a genotype C HBeAg-positive asymptomatic carrier (ASC) patient as previously described (10). The DNA sequence information from this study will appear at GenBank (accession numbers AB531977 and AB531978). To make pCR2.1-HBeAg(+), the PCR product was cloned into pCR2.1-TOPO vector (Invitrogen, Carlsbad, CA). Using the Quickchange II site-directed mutagenesis kit (Stratagene), precore stop codon mutant G1896A was induced into pCR2.1-HBeAg(+) to pCR2.1-HBeAg(−) according to the manufacturer's instructions. To obtain the mammalian cell expression vectors, we performed subcloning using the EcoRI site of pCXN2 (kindly provided by Prof. J. Miyazaki, Osaka University, Osaka, Japan), a mammalian expression vector with a β-actin-based CAG promoter and SV40 origin (28). The constructs pCXN2-HBeAg(+) and pCXN2-HBeAg(−) were generated by this method (Fig. 1A). All sequences of these plasmids were confirmed using Big Dye Terminator on a 3730 DNA sequencer (Applied Biosystems, Foster City, CA).
Cell culture
Human hepatoma cells, HepG2 and Huh7 cells, were cultured in Dulbecco's modified Eagle's medium (DMEM) (Sigma-Aldrich, St. Louis, MO) supplemented with 10% fetal bovine serum (FBS) at 37°C and 5% CO2. Approximately 1 × 105 HepG2 cells were placed on 35-mm tissue culture dishes (Iwaki Glass, Tokyo, Japan) 24 h prior to transfection (13). The cells were transfected with pCXN2-HBeAg(+) or pCXN2-HBeAg(−) in Effectene transfection reagent (Qiagen, Hilden, Germany). After 48 h, G418 was added at 1000 μg/mL for the selection of stable cell lines, and HBeAg-positive and HBeAg-negative HepG2 cells were designated. After 3 wk, to avoid monoclonal selection, all cells were collected for further analysis.
RNA extraction, cDNA synthesis, and real-time PCR
The cells were seeded into 6-well plates, and total cellular RNA was extracted 48 h later using the RNeasy Mini Kit (Qiagen) according to the manufacturer's instructions. The RNA samples were then stored at −80°C until use. RNA quality was examined using the A280/A260 ratio (Pharmacia Biotech, Bedford, MA). cDNA synthesis was performed using a random hexamer. For RNA quantitation, real-time PCR was conducted using SyBr Green I (ABI PRISM 7300; Applied Biosystems). The housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used for normalization, and data were analyzed by the comparative threshold cycle (CT) method (16). The primers used are shown in Table 1.
Real-time PCR arrays
Gene expression profiling for TLR target genes was performed using RT2 profiler PCR arrays (SuperArray, Frederick, MD) according to the manufacturer's instructions. In brief, 1 μg RNA was reverse-transcribed with the RT2 profiler PCR array first-strand synthesis assay (SuperArray), followed by real-time PCR with RT2 real-time PCR master mix SyBr green (SuperArray). Gene expression was normalized to two internal controls (GAPDH and β-actin), to determine the fold change in gene expression between the test sample (HBeAg-positive HepG2) and the control sample (HBeAg-negative HepG2) by the 2−ddCT (comparative cycle threshold) method (17). Data were analyzed with RT2 Prolifer™ PCR Array Data Analysis software (
Transfection and reporter assay
Approximately 1 × 105 cells were placed on 6-well plates (Iwaki Glass) 24 h prior to transfection. Cells were transfected with 0.4 μg of plasmid pIFN-β-luc or pNF-κB-luc in Effectene (Qiagen). For luciferase assay of NF-κB activation, cells were treated for 4 h with 0.5 or 5 ng/mL TNF-α, 10 or 50 μg/mL TLR4 ligand:lipopolysaccharide (LPS), or none at 44 h post-transfection (22,31,34,35,40). For IFN-β promoter assay, 50 μg/mL TLR3 ligand:poly(I-C), or none was added to cell culture fluid at 32 h post-transfection (16). At 48 h post-transfection, the cells were lysed with reporter lysis buffer (Promega, Madison, WI), and luciferase activity was determined by luminometer (Luminescencer-JNR II AB-2300; ATTO Bio Instruments, Tokyo, Japan) as previously described (16). Relative luciferase activity was measured at 48 h post-transfection and compared with that of an untreated control. Relative luciferase activity of HBeAg-negative cells was set as 1.
Chemiluminescent enzyme immunoassay
The supernatants of these cell lines were used for measuring the levels of HBeAg by the chemiluminescent enzyme immunoassay (CLEIA) system (Fujirebio Inc., Tokyo, Japan).
ELISA
Cell culture fluid was analyzed for IL-6 by enzyme-linked immunosorbent assay (ELISA; KOMA Biotech Inc., Seoul, Korea) following the manufacturer's protocol. Briefly, cell culture fluid samples were incubated in plates at 4°C overnight, followed by incubation with biotinylated monoclonal antibodies. Avidin-conjugated peroxidase was added to the plates, and enzyme activity was detected with an ELISA plate reader.
MTS assay
MTS assays were performed with the CellTiter 96 AQ One Solution Cell Proliferation Assay (Promega) (15). Twenty microliters/well of the MTS reagent was added to 100 μL of media containing cells in each well of 96-well plates, and left for 4 h at 37°C in a humidified 5% CO2 atmosphere. For analysis, absorbance at 490 nm was measured using a Bio-Rad iMark microplate reader (Bio-Rad, Hercules, CA).
Antiviral assay using HCV subgenomic replicon
Huh7 cells harboring HCV genotype 1b subgenomic replicon, termed C13-3 cells, were used for antiviral bioassay (14). Intracellular HCV subgenomic RNA was measured by real-time RT-PCR. C13-3 cells were incubated in cell culture supernatant from HBeAg-positive, HBeAg-negative HepG2, or control HepG2 cells for 24–48 h. Post-incubation, RNA was extracted and stored at −80°C until analysis.
Statistical analysis
Results were expressed as mean ± SD. Student's t-test was used to determine statistical significance.
Results
Detection of stable expression of HBeAg by CLEIA
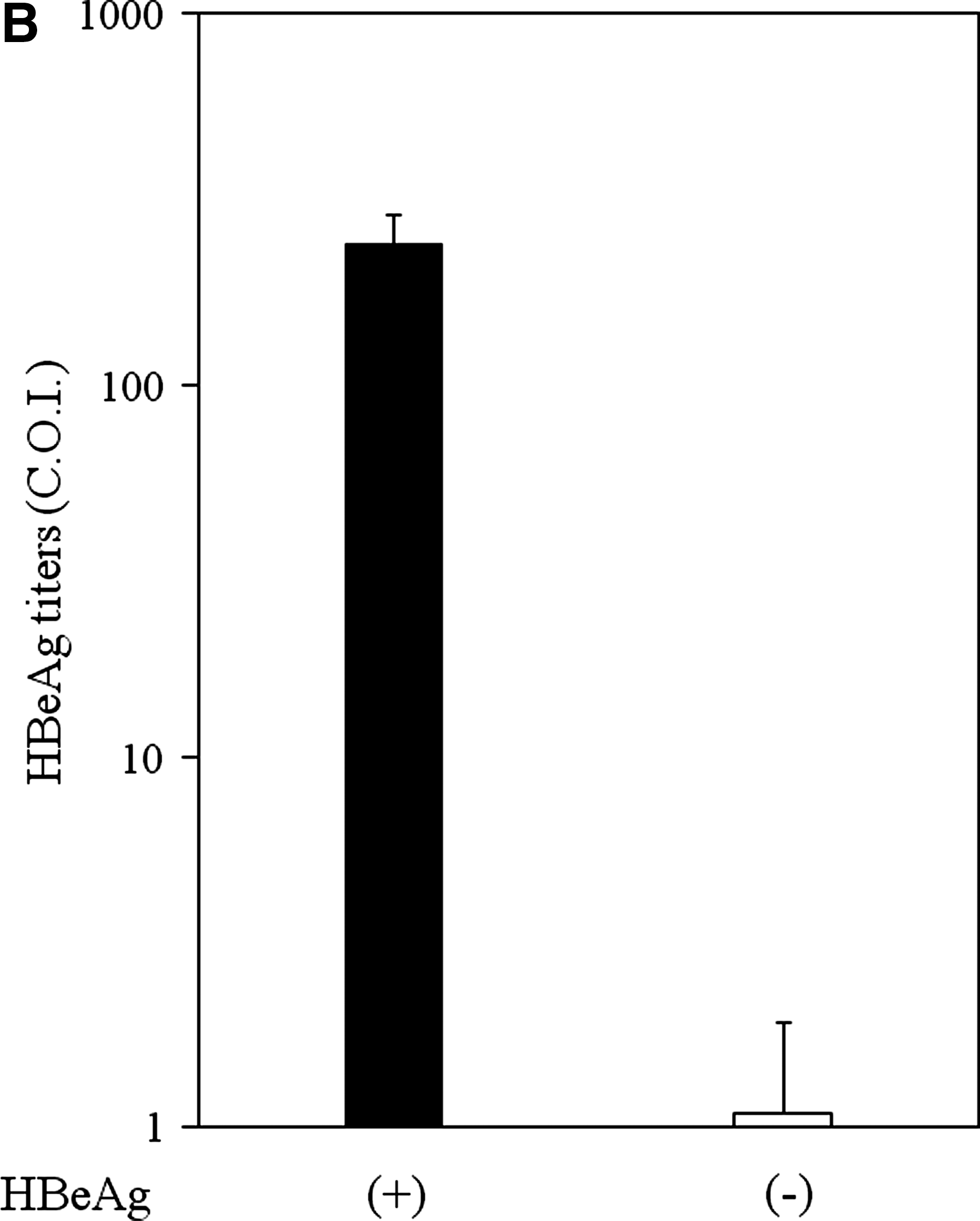
First, we examined the HBeAg production in cell culture fluid in HepG2 stably expressing HBV precore and core regions. HBeAg was detected in cell culture supernatants of HBV precore and core region-expressing cells (HBeAg-positive HepG2, 241 ± 47.9 C.O.I.) by CLEIA (cut-off index [C.O.I.]). On the other hand, expression of the core region without precore did not produce HBeAg in cell culture fluid (HBeAg-negative HepG2, 1.1 ± 0.84 C.O.I.) (Fig. 1B). Next, we performed an MTS assay to examine whether HBeAg affected cell proliferation or cell viability in our system. Cell proliferation/viability of HBeAg-positive cells (100 ± 0.87% at 24 h [n = 4]; 98.5 ± 0.7% at 48 h [n = 4]) was not statistically different from that of HBeAg-negative HepG2 (100 ± 0.4% at 24 h [n = 4]; 100 ± 1.21% at 48 h [n = 4]).
HepG2 cells respond to TLR3 ligand, TLR4 ligand, and tumor necrosis factor
Next we examined whether human hepatoma cell lines HepG2 and Huh7 respond to TLR3 ligand, TLR4 ligand, and tumor necrosis factor (TNF). Here we examined the NF-κB- and IFN-signaling pathways in HepG2 and Huh7 cells. To examine whether HepG2 possesses a functional TLR4 pathway, we initially characterized LPS-induced activation of NF-κB in HepG2 and Huh7 by luciferase reporter assay. TLR4 plays an important role in the activation of NF-κB following exposure to extracellular LPS. When LPS was added to the cell culture medium of HepG2 and Huh7, approximately 23∼56-fold and 1.8∼3.0-fold activation, respectively, of NF-κB activity were observed (Table 2). Similarly, when TNF-α, another NF-κB activator, was added to the cell culture medium of HepG2 and Huh7, respectively, approximately 9∼14-fold and 1.4∼8.6-fold activation, respectively, of NF-κB activity were observed (Table 2). However, to examine for a functional TLR3 pathway by luciferase reporter assay, when poly(I-C) was added to the cell culture medium of HepG2 and Huh7, respectively, approximately 1.69-fold and 0.93-fold activation, respectively, of IFN-β-promoter activity were observed (Table 2), supporting the view that Huh7 cells are defective in the TLR3 and RIG-I pathway (16,39). Our results suggested that HepG2 possesses functional TLR3 and TLR4 pathways to some extent, but Huh7 does not possess a functional TLR3 pathway.
Cells were transfected with 0.4 μg of plasmid pIFN-β-luc or pNF-κB-luc in Effectene (Qiagen). For the luciferase assay of NF-κB activation, cells were treated for 4 h with 0.5 or 5 ng/mL TNF-α, 10 or 50 μg/mL LPS, or none, at 44 h post-transfection (22,31,34,35,40). For the IFN-β promoter assay, 50 μg/mL poly(I-C) or none was added to cell culture fluid at 32 h post-transfection (16). Relative luciferase activity was measured at 48 h post-transfection and compared with that of an untreated control. Results are expressed as mean ± SD.
*p < 0.01, **p < 0.001, and ***p < 0.0001 in HepG2 or Huh7 induced by each ligand compared with untreated controls.
Downregulation of IFN and cytokine gene expression by HBeAg
Since HBeAg is associated with immune tolerance (3,42), we wanted to determine whether this might be related to HBeAg suppressing the host innate response, including the production of cytokines. To confirm the downregulation of IFN and cytokine genes, we performed real-time RT-PCR assays. We compared six IFN and cytokine (IFN-α1, IFN-β, IL-6, IL-8, IL-12A, and TNF) gene expressions in HBeAg-positive HepG2 cells with those in HBeAg-negative HepG2 cells. The mRNAs of IFN-α1 and IL-12A were inhibited (6.7% and 11.6%, respectively, of those in HBeAg-negative HepG2), and ΔCt of HBeAg-positive HepG2/ΔCt of HBeAg-negative HepG2 in IFN-α1 mRNA and those in IL-12A mRNA were 14.36 ± 0.11/10.47 ± 0.02 (p < 0.001, n = 3), and 17.74 ± 0.11/14.65 ± 0.17 (p < 0.001, n = 3), respectively. As shown in Fig. 2, more inhibition of IFN-β, IL-6, IL-8, and TNF mRNA in HBeAg-positive HepG2 were also observed, compared with HBeAg-negative HepG2.
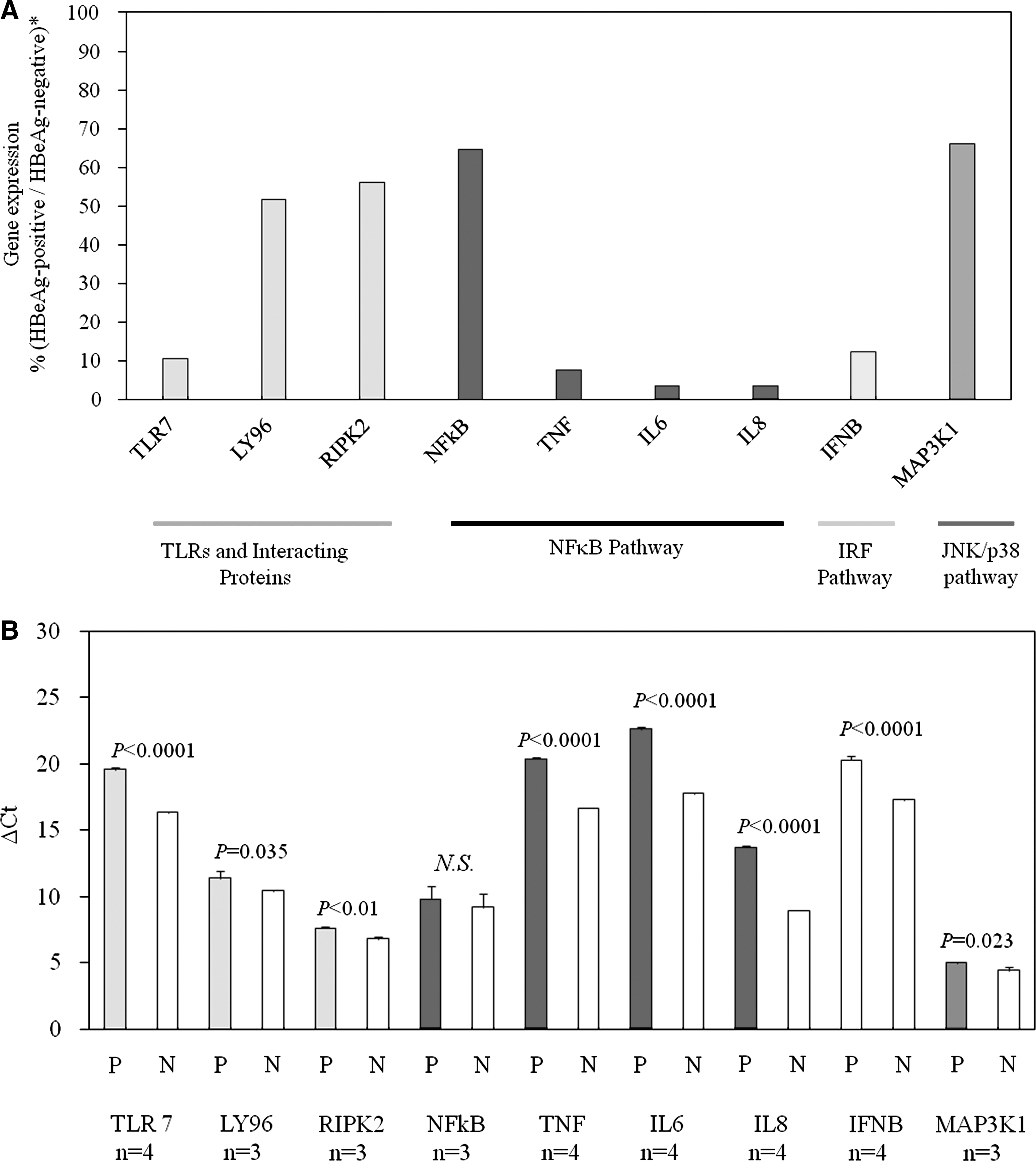
Effects of HBeAg on toll-like receptor (TLR) signaling-related gene expression (comparison of genes expressed in HBeAg-positive HepG2 with those in HBeAg-negative HepG2). (
Effects of TLR-dependent target gene expression by HBeAg
To explore the upstream mechanism of IFN and cytokine production, we performed RT2 profiler array assays to analyze important TLR-activated genes (84 target genes were included in the RT2 profiler array), that could be modulated by TLR-signaling, from HBeAg-positive HepG2. Expression profiling showed more than twofold inhibition of nine genes compared to that of HBeAg-negative HepG2 cells (TLR7, LY96, RIPK2, NFκB1, TNF, IL-6, IL-8, IFN-β, and MAP3K1) (Fig. 2A). To confirm these results, real-time PCR was performed. All of these genes except NF-κB1 were significantly downregulated in HBeAg-positive cells compared to HBeAg-negative cells. All of these genes have important roles in the immune response and activation of transcription (Fig. 2B).
Effects of HBeAg on NF-κB activation
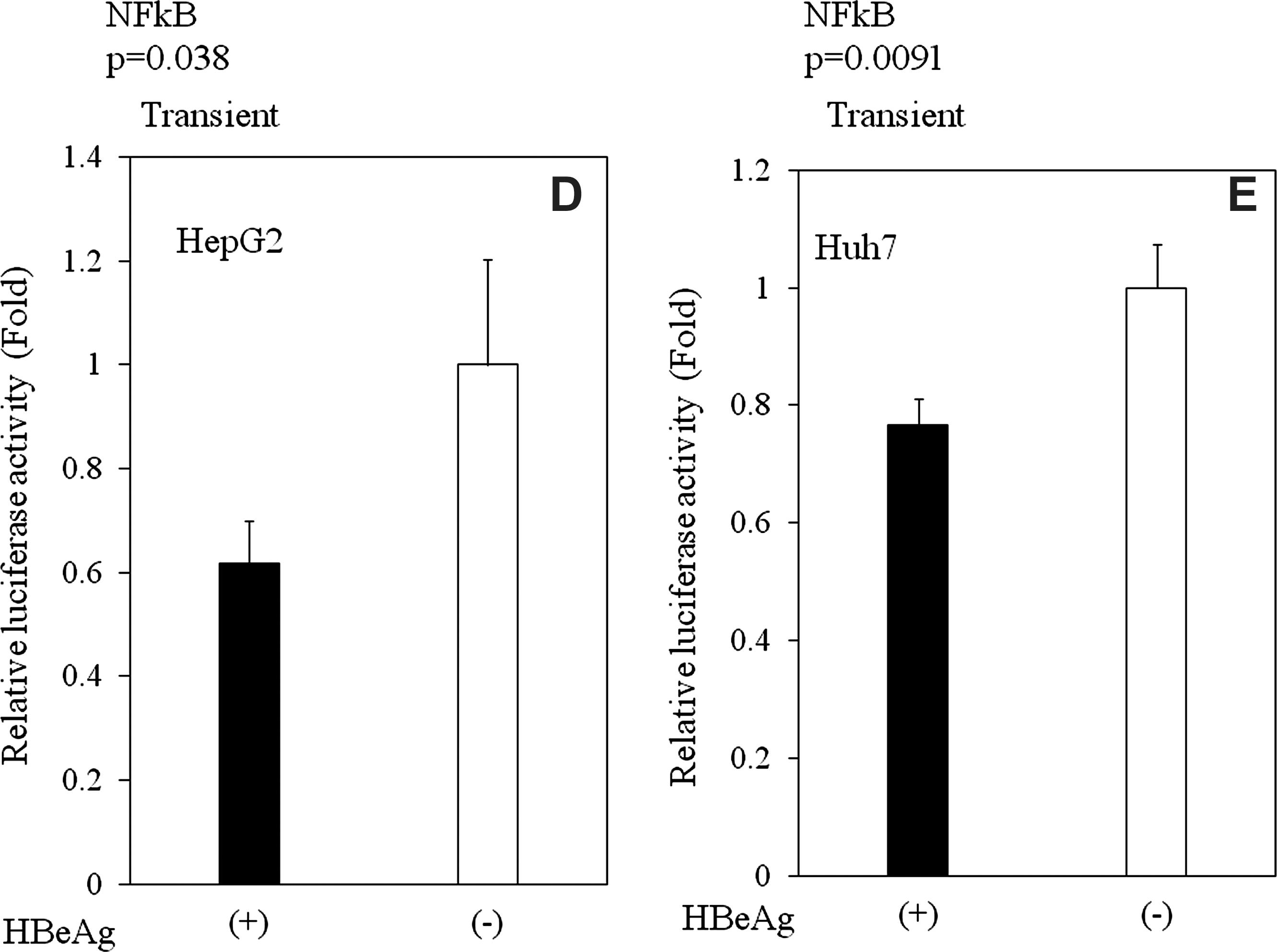
Next, we assessed the mechanisms by which HBeAg affects cytokine and IFN production. HBV activates NF-κB, a major player in innate immune responses to viral infections (19). Therefore, we postulated that HBeAg inhibition of the activation of NF-κB might result in the inhibition of cytokine and IFN production, and the subsequent escape of an antiviral response. To test this assumption, we expressed luciferase reporter protein under the control of an NF-κB-dependent promoter in HBeAg-positive or HBeAg-negative HepG2 with or without TNF-α or LPS stimulation (Fig. 3A–E). As expected, HBeAg inhibited NF-κB promoter activity in HBeAg-positive HepG2 cells (Fig. 3A–C; p < 0.001 with no drug [n = 3]; p = 0.004 with LPS [n = 3]; p = 0.0075 with TNF [n = 3]). These results were also confirmed by the transient HBeAg-expression assay in HepG2 (p = 0.038, n = 3) and Huh7 cells (p = 0.0091, n = 3) (Fig. 3D and E). These findings suggest that HBeAg may affect cytokine production, at least in part, through NF-κB.
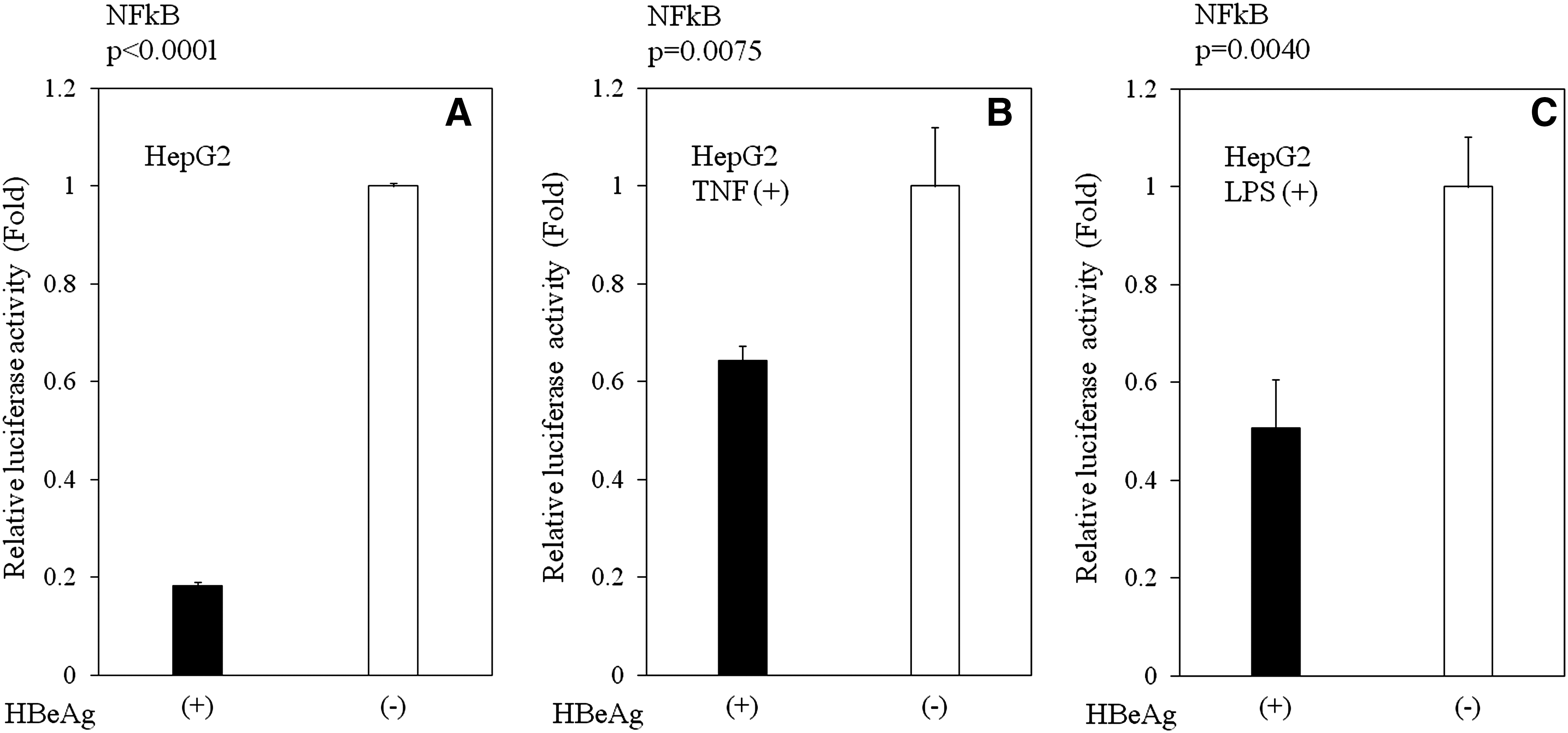
Effects of HBeAg expression on the activation of NFκB-dependent promoters. Plasmids expressing luciferase under the control of NFκB-dependent promoters were transfected into HBeAg-positive or HBeAg-negative HepG2 cells. At 44 h post-transfection, none (
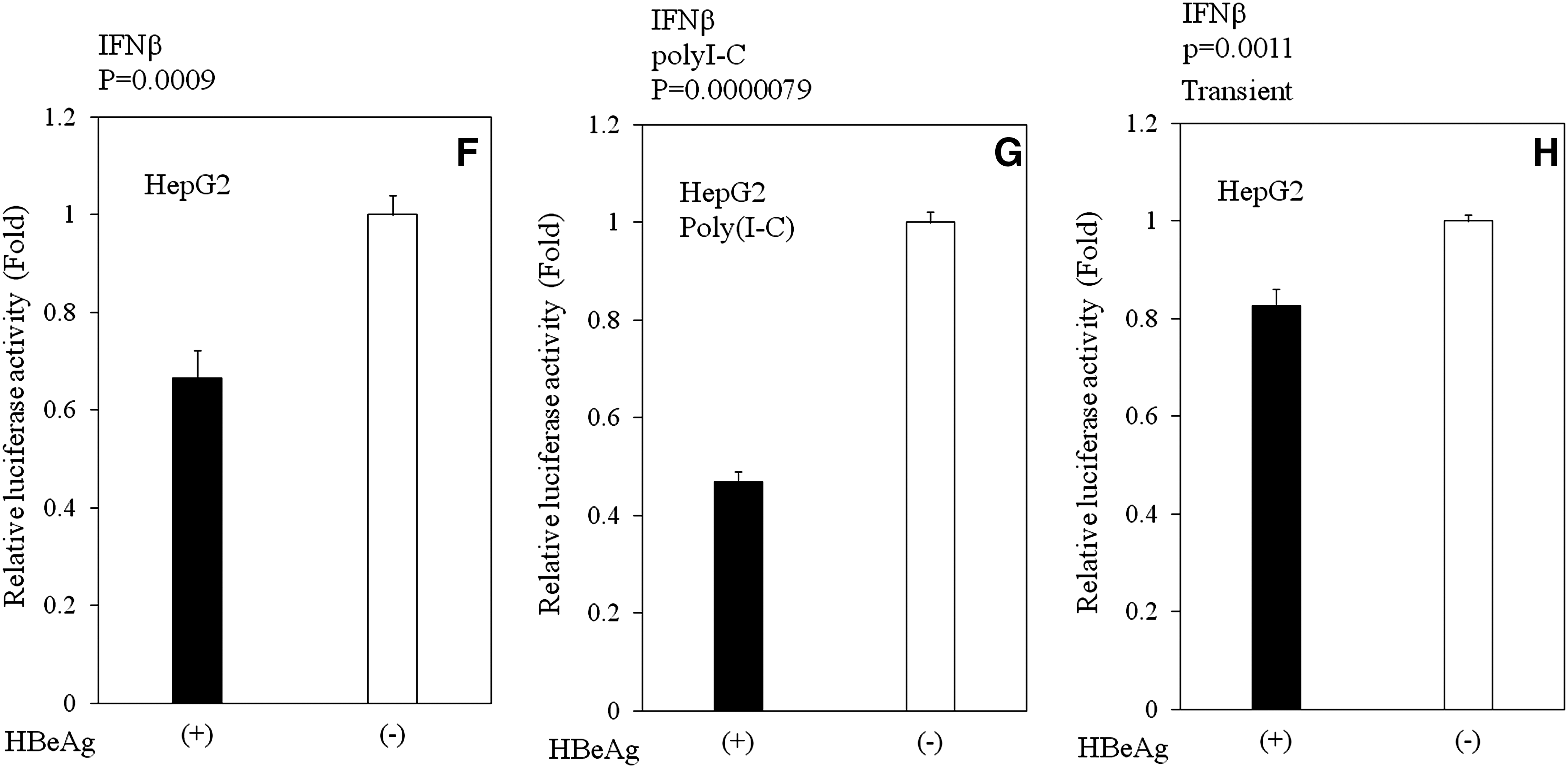
Effects of HBeAg on IFN-β activation
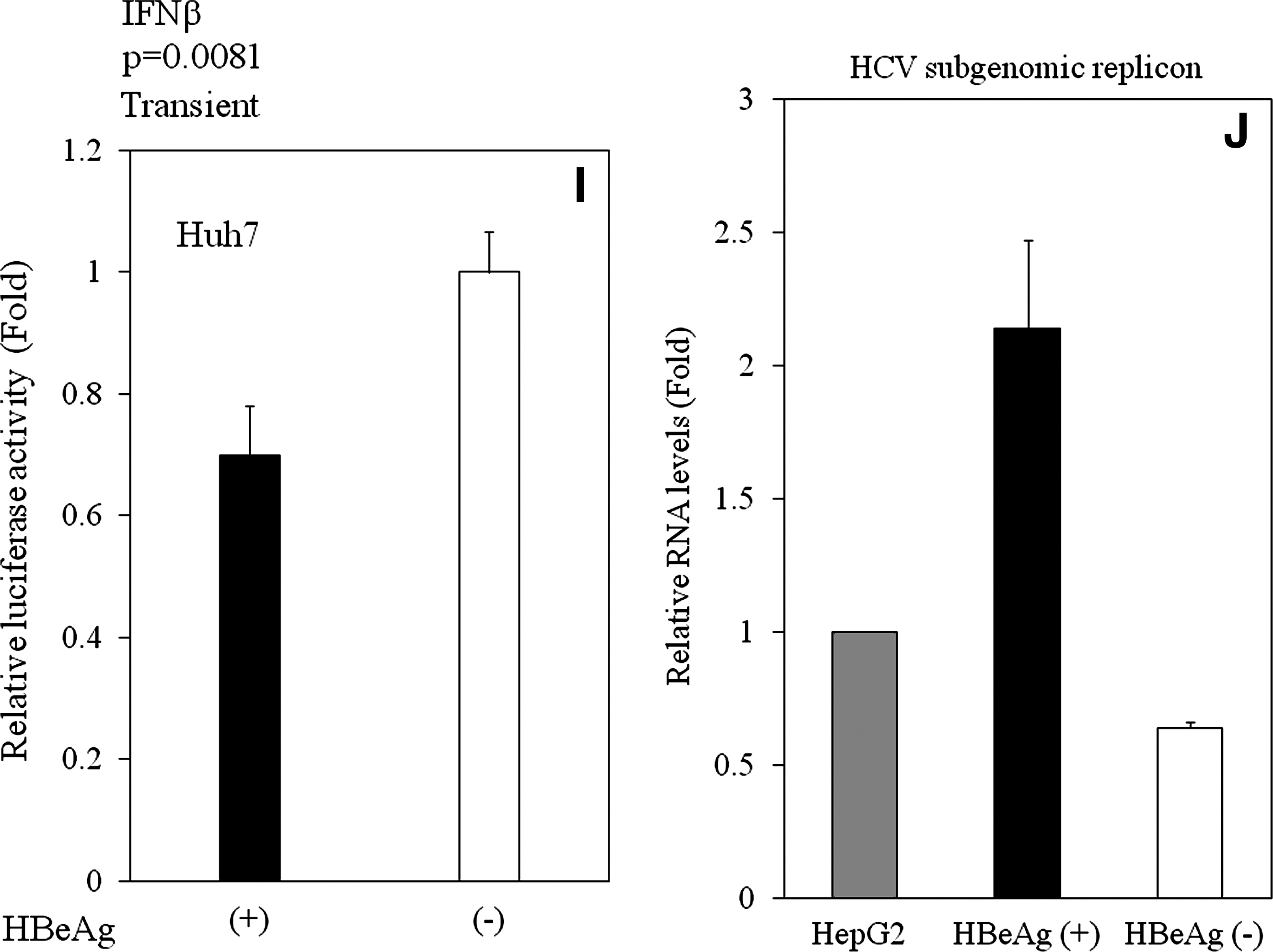
NF-κB stimulation leads to the expression of multiple cellular factors, including IFN-β, a central player in the innate immune response that is activated upon virus infection. In order to ascertain whether HBeAg inhibits IFN-β-promoters, we performed experiments using IFN-β-promoter luciferase reporter, essentially as described in the previous section. That is, we used the luciferase gene under the control of an IFN-β-stimulated promoter, and examined its expression in HBeAg-positive and HBeAg-negative HepG2 cells. HBeAg inhibited IFN-β-stimulated promoter activity in HBeAg-positive HepG2 cells with (p < 0.001, n = 3), or without poly(I-C) (p < 0.001, n = 3) (Fig. 3F and G). We also confirmed these results by transient transfection experiments with HepG2 (p = 0.0011, n = 3), and with Huh7 (p = 0.0081, n = 3) (Fig. 3H and I). These results demonstrated that HBeAg inhibits both NF-κB- and IFN-β-signaling pathways in hepatocytes.
Cell culture fluid from HBeAg-positive HepG2 enhanced HCV subgenomic RNA replication
To confirm the function of IFN production of these cell lines, we examined whether conditioned media from HBeAg-positive or HBeAg-negative HepG2 cells would cause any differences in HCV subgenomic RNA replication, which is IFN-sensitive replication (14), as it has been reported that there are no direct interactions between HBV and HCV replication in cell culture models and in a mouse study (1,9,11). Cell culture fluid from HBeAg-positive HepG2 cells enhanced HCV subgenomic RNA replication, more than that from HBeAg-negative HepG2 cells (Fig. 3J; p = 0.0014, n = 3), suggesting that HBeAg-expressing HepG2 cells contain less IFN than do HBeAg-negative cells, and that conditioned medium from HBeAg-positive HepG2 cells contains less IFN than that from HBeAg-negative cells. In this system, when we treated cells with 0, 1, 10, 100, and 1000 U/mL IFN-α, HCV subgenomic RNA levels were 100%, 57%, 39%, 28%, and 25%, respectively. We estimated that conditioned medium from HBeAg-negative HepG2 cells was equal to ∼10 IU/mL IFN-α. Our results showed that HBeAg inhibits IFN production in cell culture medium.
Since the NF-κB target gene IL-6 has also been implicated in hepatitis B pathogenesis (30), the modulation of IL-6 involved in innate signaling by HBeAg was also verified at the protein level by ELISA. Our results demonstrated that IL-6 expression was downregulated in HBeAg-positive HepG2 cells (36.6 ± 30.1 pg/mL; 0 ± 0 pg/mL in conditioned medium from HepG2 control cells; 324.2 ± 15 pg/mL in conditioned medium from HBeAg-negative HepG2 cells). The concentration of IL-6 from HBeAg-positive HepG2 cells was significantly lower than that from HBeAg-negative HepG2 cells (p = 0.00012, n = 3).
Discussion
In this study, we investigated the regulation of HBeAg-induced suppression of IFN and cytokines in HepG2 stably expressing HBeAg protein as a model cell line. Our results demonstrated that HBeAg expression inhibits IFN and cytokine production. Transient expression of HBeAg also downregulated both NF-κB- and IFN-β-promoter activity in HepG2 or Huh7, although the mechanisms for this downregulation are unknown. In contrast to our findings, Yang et al. (46) observed that HBeAg activates NF-κB through IκBα degradation, and produces TNF-α and GM-CSF in the human hepatoma cell HA22T/VGH. These differences between their findings and ours may have been caused by the differences in the cell lines, and/or promoters (33). Extensive immunological studies by the Milich group (3,4,27) demonstrated that HBeAg appears more efficient at eliciting T-cell tolerance, including production of its specific cytokines IL-2 and IFN-γ, than HBV core antigen. Our observations support the immune-modulating role of HBeAg.
Locarnini et al. (23) used the Tet-off tetracycline gene expression system in Huh7, and revealed that core/precore expression affected gene expression, including cytokines. The system used in our present study, with HepG2 stably expressing HBeAg, supports these findings. Our results provide further direct evidence that hepatocytes exposed to HBeAg have enhanced HCV subgenomic RNA replication, and are significantly influenced in their ability to replicate. Several recent reports have also suggested that there was no evidence of direct interaction between HBV and HCV (1,9,11), although clinical studies showed interaction between HBV and HCV replication (24). It is possible that HBV might interfere with another virus by IFN or another cytokine. A cytokine response is critical for clearance of HCV, as failure to mount a potent and broad T-cell-repertoire response results in persistent HCV replication. This would explain how patients dual-infected with HBV and HCV exhibit a selective deficit of anti-HCV immunity, while demonstrating preservation of a normal immune response to unrelated antigens.
We used RT-PCR to observe the expression of TLRs 1, 3, 4, 5, 6, and 7 in HepG2 cells. We also confirmed in the present study that HepG2 has functional TLRs 3 and 4. Preiss et al. (32) could not detect an NF-κB response to 1 ng/mL–1 μg/mL LPS in HepG2, whereas we could detect such a response to 10–50 μg/mL LPS (Table 2). Downregulation of TLR2 mRNA by genotype C HBV-derived HBeAg was not observed in our study, in contrast to the results of a previous study (43), in which genotype D HBV-derived clone (23) was used. Xu et al. (45) reported that TLR7 was suppressed in HBV infection, supporting our results. We do not know why LY96, an important molecule for TLR4, is downregulated (Fig. 2). Viruses encode proteins that target various intracellular signaling pathways, causing their constitutive or prolonged activation, resulting in increased cell proliferation and survival (41). It is well known that HBV activates the MAPK pathway (5). It is also known that RIPK2 activates the NF-κB- and IFN-β-dependent antiviral responses (8). These findings were in accordance with HBeAg's inhibition of the production of IFNs and cytokines (Fig. 2).
What is the mechanism of the downregulation of cytokine production by HBeAg? From our results (Fig. 2), HBeAg appears to interact with the TLR signaling pathway upstream of NF-κB. In LPS stimulation, we observed downregulated TLR4 in HBeAg-positive HepG2 cells (data not shown). Although we are currently investigating this issue, TLR4 might be one of the more important molecules. Precore protein also may affect intracellular signal transduction pathways. Further studies will be needed to clear up these issues.
Many viruses have evolved strategies that block the effector mechanisms induced through IFN- and/or cytokine-signaling pathways (17). Although multiple mechanisms contribute to viral persistence, the ability of the virus to evade innate immune responses is likely to be particularly important. In this report, we have demonstrated that HBeAg suppresses IFN and cytokine mRNA expression. Exploration of the novel HBeAg-inhibiting signaling pathways could lead to the development of new therapeutic strategies for persistent HBV infection.
Footnotes
Acknowledgements
We are grateful to Satomi Hasegawa for excellent technical assistance, and Prof. Junichi Miyazaki and Dr. Naoya Kato for providing the materials. This work was supported by grants for Scientific Research 15790338, 21590829, 21590828, and 21390225, from the Ministry of Education, Culture, Sports, Science and Technology, Japan (T.K., F.I., and O.Y.), a grant from the Ministry of Health, Labour and Welfare of Japan (O.Y.), a Special Coordination Fund for Promoting Science and Technology of the Ministry of Education, Culture, Sports, Science and Technology, of the Japanese Government (T.K.), and a grant from the Global Centers of Excellence Program at Chiba University (S.W.).
Author Disclosure Statement
The authors have no conflicts with regard to financial interests. This material has not been previously reported and is not under consideration for publication elsewhere.