
Abstract
The replication of vesicular stomatitis virus (VSV) in isolated human leukocytes has been used to measure the level of nonspecific antiviral immunity. However, during infection with some pathogens, the main effect observed is caused by interaction between the pathogen and VSV. This was also noted in advanced stages of HIV infection, when an inverse association between HIV viral load and VSV replication was found. The mutual effect was markedly stronger than the correlation between the VSV replication level and CD4+ T-cell count. Since successful antiretroviral therapy is associated with a decrease in HIV viremia to undetectable levels, the effect of such therapy on VSV replication was expected and confirmed in this investigation. In fact, increased VSV titers were observed together with decreased HIV viral load, particularly in the case of efficient therapeutic schemes, for example those including lopinavir/ritonavir. The results showed that VSV replication capacity reflected the progression of HIV infection. Moreover, the presence of interferon in the plasma of AIDS patients was found to be only partially responsible for the inhibition of VSV replication. The results suggest a specific HIV-VSV interaction, whether direct or indirect. Thus the VSV replication assay may be applied in evaluating the stage of HIV infection.
Introduction
To determine the innate antiviral immunity level in peripheral blood leukocytes (PBLs), an assay using vesicular stomatitis virus (VSV) was developed (3,4,15,19,21,25). VSV was chosen for use in the immunity analysis of Europeans because it does not establish natural infections in Europe (23). Because of this the development of VSV infection is believed to be suitable for primary infection, and only nonspecific immune responses can inhibit replication of the virus. The measurement of VSV replication has thus been applied to characterize innate antiviral immunity (3,4,15,19,21,25). Comparison of the leukocytes of healthy people with those of patients suffering from recurrent infections of the upper respiratory tract showed that VSV replication reached significantly higher titers in the latter group (15). Similar results were obtained in acute leukemia patients (4), but no differences were found in a group of hemodialysis patients with chronic renal failure or in HCV-positive persons (25).
The findings obtained with the VSV replication assay also depend on factors unconnected with disease progression. Age-related differences were observed which showed reduced innate immunity in persons over 60 years of age, particularly in men (19). Medicines that interact with immune functions could change the natural resistance of human leukocytes to viral infection. Such effects were observed after application of baicalein (3) and donepezil (21), which augmented the resistance of PBLs to VSV. These factors should be taken into consideration during the analysis of the results.
Similarly, the course of HIV infection is affected by many miscellaneous factors, both host-dependent (e.g., CCR5-Δ32 mutation resulting in insensitivity to infection) (20), and virus-dependent (e.g., HIV strain variations resulting in drug-resistant disease). The impaired effectiveness of innate immunity caused by dysfunction or weakness of certain components of the response may also play important roles. One of the symptoms of such impairment is connected with the interferon system. High levels of atypical acid-labile interferon-α are seen in the plasma of AIDS patients (11,17,18,22). Macrophage activities, including phagocytosis, intracellular killing, chemotaxis, and cytokine production, are also severely impaired during the course of infection (10).
The aim of this study was to ascertain whether the VSV replication assay could reflect the progression of HIV infection and the effectiveness of applied therapy. The study focused on determining which progression marker of HIV infection most strongly correlated with VSV replication.
Materials and Methods
Subjects
The study participants included 66 HIV-1-positive patients of the Department of Infectious Diseases of Wroclaw Medical University. At study entry, serological tests (EIA) for anti-HCV and anti-HBc antibodies and HBs antigen were performed once. The control group consisted of 32 healthy individuals with no HIV, HBV, or HCV infection history. Informed consent was obtained from the patients (Table 1).
ART, antiretroviral therapy; IDU, intravenous drug user; HCV, hepatitis C virus; HBV, hepatitis B virus; HIV, human immunodeficiency virus.
Antiretroviral therapy
Combination antiretroviral therapy (ART) was initiated according to current recommendations. Two groups of patients on ART were compared: (1) backbone therapy including lopinavir/ritonavir (LPV/RTV), and (2) backbone therapy and another protease or non-nucleoside inhibitor.
Quantification of HIV-1 RNA in plasma
Plasma HIV-1 RNA was measured in −70°C plasma samples prepared from blood using the Cobas Amplicor HIV-1 Monitor v. 1.5 (Roche Diagnostics Corp., Indianapolis, IN) standard assay (lower limit of detection: 400 copies/mL), or ultrasensitive assay (lower limit of detection: 50 copies/mL).
CD4+ T-cell count
Absolute CD4+ T-cell counts were determined by flow cytometry with a FACScan device (BD Biosciences, San Jose, CA).
Innate immunity assay
Leukocytes were isolated from heparinized peripheral blood (heparin without preservatives at a concentration of 10 U/mL of blood) by gradient centrifugation in a Gradisol G device (Aqua Medica, Poland), with a density of 1.115 g/mL and a concentration of 2 × 106 cells/mL (15). Vesicular stomatitis virus (VSV Indiana strain, Rhabdoviridae) was multiplied and titrated in L929 cells (ATCC CCL1). The titer of virus was expressed with reference to the value of tissue culture infectious dose (TCID50), based on the cytopathic effect induced by the virus in ∼50% of cells in the tissue culture. Leukocytes (2 × 106/mL) were infected with VSV at a concentration of 100 TCID50/mL. After 40 min of adsorption at room temperature, the virus was washed out three times with 5 mL of RPMI with 2% calf serum. A sample of infected cells was set aside and kept at a temperature of 4°C to serve as a control of the starting level of the virus. The other cells were incubated at 37°C, and samples of medium above the infected cells were collected each day and titered into L929 cells. The titer of virus was expressed in units of TCID50 on the logarithmic scale.
Replication of VSV was measured in PBL cultures. The virus titers were determined 24, 48, and 72 h after VSV infection. The highest titer obtained was analyzed as the final result of the determination. A lack of VSV replication (log TCID50/mL = 0 or 1) showed a high level of innate antiviral immunity or the presence of an interfering factor. High VSV titers suggested weak innate immunity.
Simultaneously with infection with VSV, the leukocyte cultures were stimulated with 10 μg/mL phytohemagglutinin (PHA; Sigma-Aldrich, St. Louis, MO) for 48 h. Interferon activity was determined in the collected supernatants. The cytokine's activity was also measured in plasma obtained from blood samples before leukocyte isolation.
Anti-VSV activity
Plasma samples or culture medium (control) was incubated with VSV (106–108 TCID50/mL) for 2 h at 37°C, then the virus titers were determined. In this study a decrease of viral titer above 1 log was regarded as a positive result.
Quantification and characterization of IFN
Confluent monolayers of A549 cells (human lung adenocarcinoma, ATCC CCL 185) were prepared on 96-well microplates in Dulbecco's modified Eagle's minimum essential medium (DMEM), with 10% calf serum, L-glutamine, and antibiotics (11,17,18). IFN samples diluted on separate plates were added to the cell monolayers and incubated at 37°C for 24 h in 5% CO2 in humidified air. The cells were then washed and challenged with 100 TCID50 of encephalomyocarditis virus (EMCV). After 48 h of incubation at 37°C, the cytopathogenic effect was determined using the modified MTT method (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide; Sigma-Aldrich), in which the cell cultures were incubated with 0.5–1.0 mg/mL MTT for 2 h at 37°C, and then SDS/DMF solution (i.e., 13.5% [w/v] sodium dodecyl sulfate in 45% [v/v] dimethylformamide; Sigma-Aldrich), was added for at least 2 h and the absorbance at 570 nm was measured. The endpoint of IFN titration (50% reduction of cell killing) was calculated. Laboratory and/or international standards (NIBSC 61 9/19 and NIAID G-023-902-527) of IFNs were included in all the assays.
Antiviral assays
Lopinavir (LPV) and ritonavir (RTV) in substance form were obtained from Abbott Laboratories.
Viricidal (anti-VSV) activity of LPV and RTV
The compounds in various concentrations (1–1000 μg/mL) were incubated with vesicular stomatitis virus (VSV, Rhabdoviridae, enveloped virus) (24). The virus was used at doses of 106–107 TCID50/mL. After 2 h of incubation at room temperature, the virus titer was determined in human A549 cells. To detect the viricidal effect, an at least 100-fold reduction of VSV titer must be found.
Effect of LPV and RTV on VSV replication
VSV was replicated in A549 cells in the presence of various concentrations (1–1000 μg/mL) of the compounds. The cultures were infected with 103–104 TCID50 VSV at room temperature. After 1 h, the supernatants (with unattached virus) were removed and the cultures were incubated for 48 h at 37°C. Then the supernatants (with replicated virus) were collected and the VSV titers were measured in A549 cells. To evaluate the decrease in viral replication, an at least 100-fold reduction of VSV titer must be found. Parallel incubation at 4°C was performed as a control under non-permissive conditions.
Results
The determination of antiviral immunity was performed in blood samples taken once or twice (in the case of changes in clinical status) from 66 HIV-infected patients and 32 individuals uninfected and not exposed to HIV (control group). The group of HIV-positive individuals consisted of patients at different stages of infection based on CD4+ T-cell counts ranging from 3–880 cells/μL. Forty-one samples were taken from patients who were not on ART, because they had planned to initiate therapy or did not require it. Sixty-five tests were performed in patients who received ART (Table 1). Most of the patients had been intravenous drug users (IDUs), but had stopped taking drugs more than 6 mo before enrollment in the study, and no additional factors influencing VSV replication were found. Although most of the IDUs were infected with HCV and had past HBV infection, no correlations between the hepatic infections and VSV replication were observed (data not shown).
Because natural VSV infection may interfere with the VSV replication assay, two procedures were introduced. First, subjects known to have visited America (from anamnesis) were excluded from the study. Second, the potential presence of specific anti-VSV activity in plasma was investigated in some patients, particularly in samples that demonstrated total inhibition of VSV replication. No samples with anti-VSV activity, suggesting previous infection with the virus, were detected (data not shown).
VSV replication in the control group (Fig. 1) was inhibited (log TCID50 ≤ 1) in 20 of the 32 persons (63%), reflecting good antiviral immunity. Only 6% showed high (not inhibited, log TCID50 ≥ 4) levels of VSV replication.
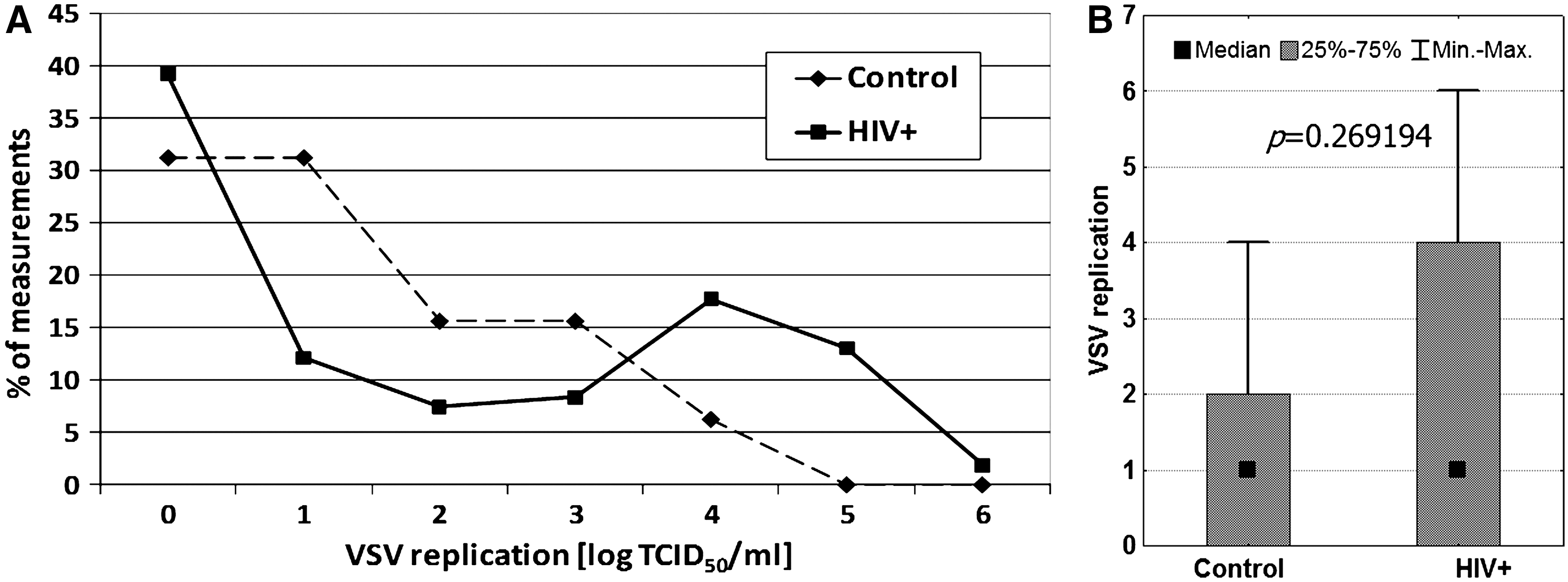
Interestingly, VSV replication formed two peaks in the HIV-infected patients (Fig. 1), revealing heterogeneity of the group. A lack of VSV replication was observed in 50% of the measurements, and high levels of VSV (log TCID50 ≥ 4) were found in 34% of the samples. There were no significant differences between healthy controls and HIV-infected patients. In fact, the HIV-positive patients had various clinical statuses (as assessed by CD4+ cell count and viral load). Moreover, antiviral treatment should also be taken into consideration, as the medications used in ART could interact with the results of innate immunity determination.
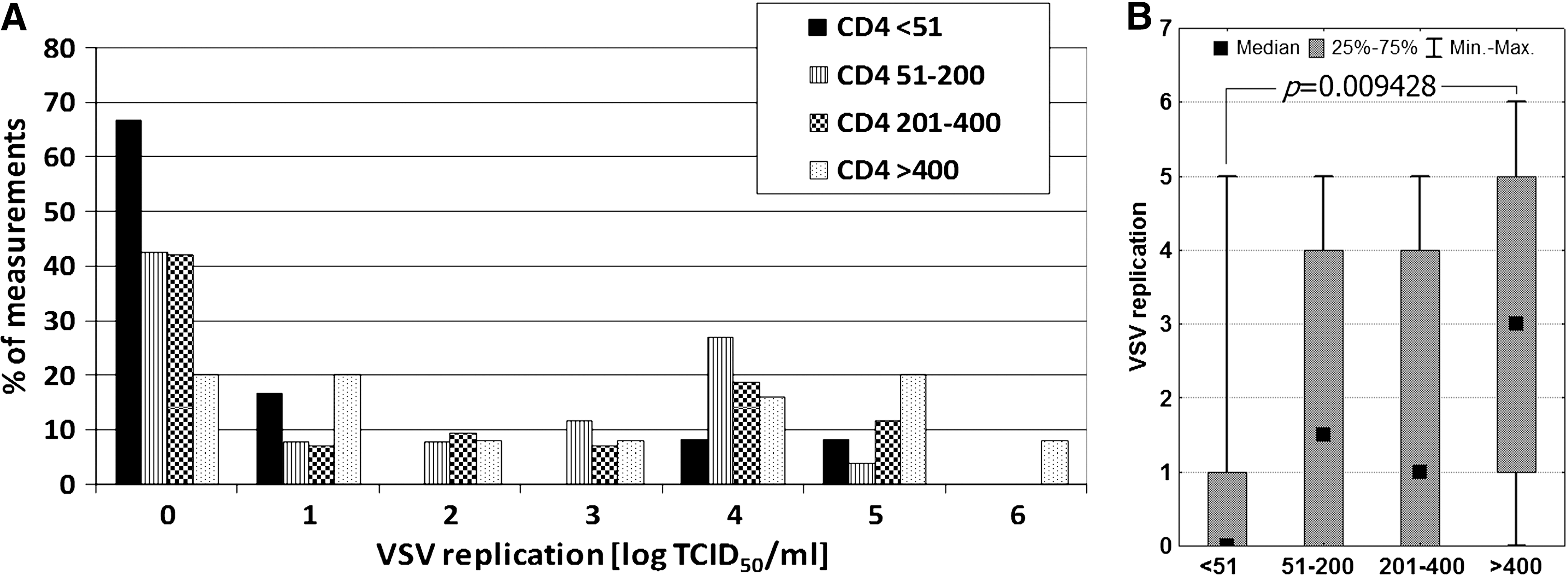
Decreased VSV replication was found to only slightly correlate with decreased CD4+ T-cell counts (Fig. 2). The mean log TCID50 VSV/mL values were 0.92, 1.85, 1.79, and 2.72, for the CD4+ T-cell count groups of <51 (n = 12), 51–200 (n = 26), 201–400 (n = 41), and >400 cells/μL (n = 25), respectively. Similar results were obtained when median values were compared. For these same groups the median values were 0, 1.50, 2, and 3, respectively. However, significant differences were found only when comparing the CD4 <51 and CD4 >400 cells/μL groups.
In contrast to CD4+ T-cell counts, the number of CD8+ T cells did not correlate with VSV replication. The mean log TCID50 VSV/mL values were 2.17, 1.47, 2.30, and 2.30 for the CD8+ T-cell count groups of <400 (n = 12), 401–800 (n = 30), 801–1200 (n = 20), and >1200 cells/μL (n = 20), respectively.
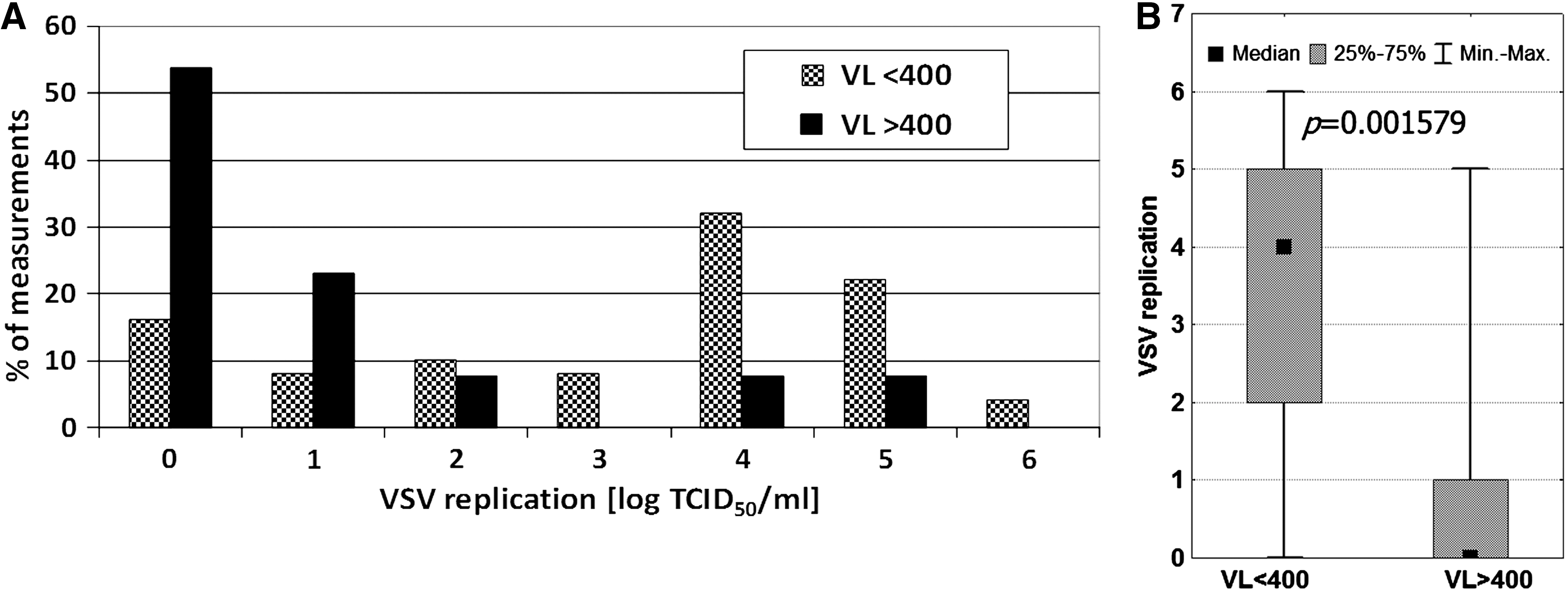
The correlation between HIV RNA level and VSV replication was also evaluated. Two groups of patients were analyzed according to viral load (VL): those with a detectable level of HIV RNA, and those with VL below the standard limit of detection (400 copies/mL) (Fig. 3). A highly significant difference was observed. The mean log TCID50 VSV/mL for samples with HIV RNA VL <400 copies/mL was 3.00, but it was only 1.13 when VL was ≥400 copies/mL. This difference was even more apparent for the median values (4 and 0, respectively). The results suggest that the leukocytes' ability to replicate VSV was inversely associated with HIV viremia. Regardless of the reason for the relationship found between HIV RNA level and VSV replication, it showed that the VSV replication assay revealed active HIV replication more than the status of antiviral activity of innate immunity. Moreover, the measured HIV and VSV levels were found to be more strongly associated with each other than with CD4 count. This is illustrated by the lack of statistical significance (p = 0.698) of the difference in CD4+ T-cell counts between samples with VL <400 copies/mL (CD4+ count: mean 329, median 287, range 6–880 cells/μL; n = 61), and those with VL >400 copies/mL (CD4+ count: mean 231, median 244, range 3–862 cells/μL; n = 17).

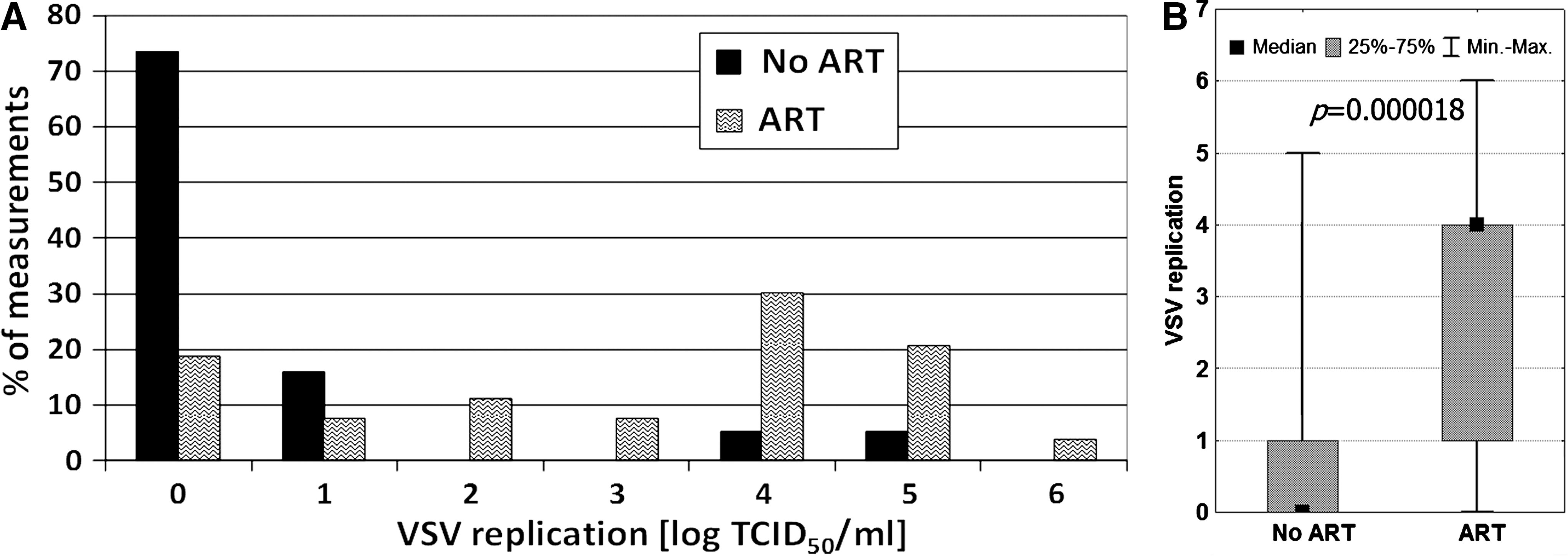
CD4+ T-cell count and VL are important markers used to assess the efficacy of ART. Since current therapy protocols are usually associated with clinical improvement, an effect on VSV replication was expected. Comparison of the results obtained for treated leukocytes of patients on ART and those not on ART revealed a very significant difference (Fig. 4). The mean log TCID50 VSV/mL for the untreated patients was 0.49, and for treated patients was 2.98. The corresponding median values were 0 and 4.

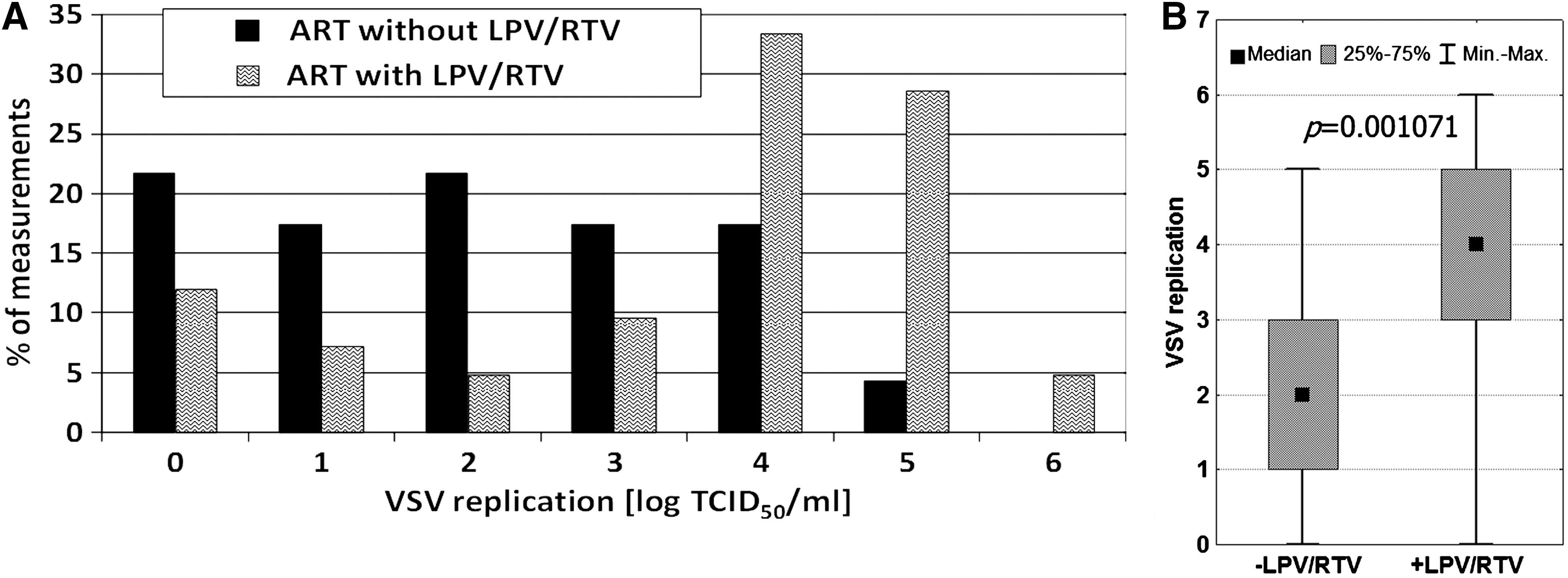
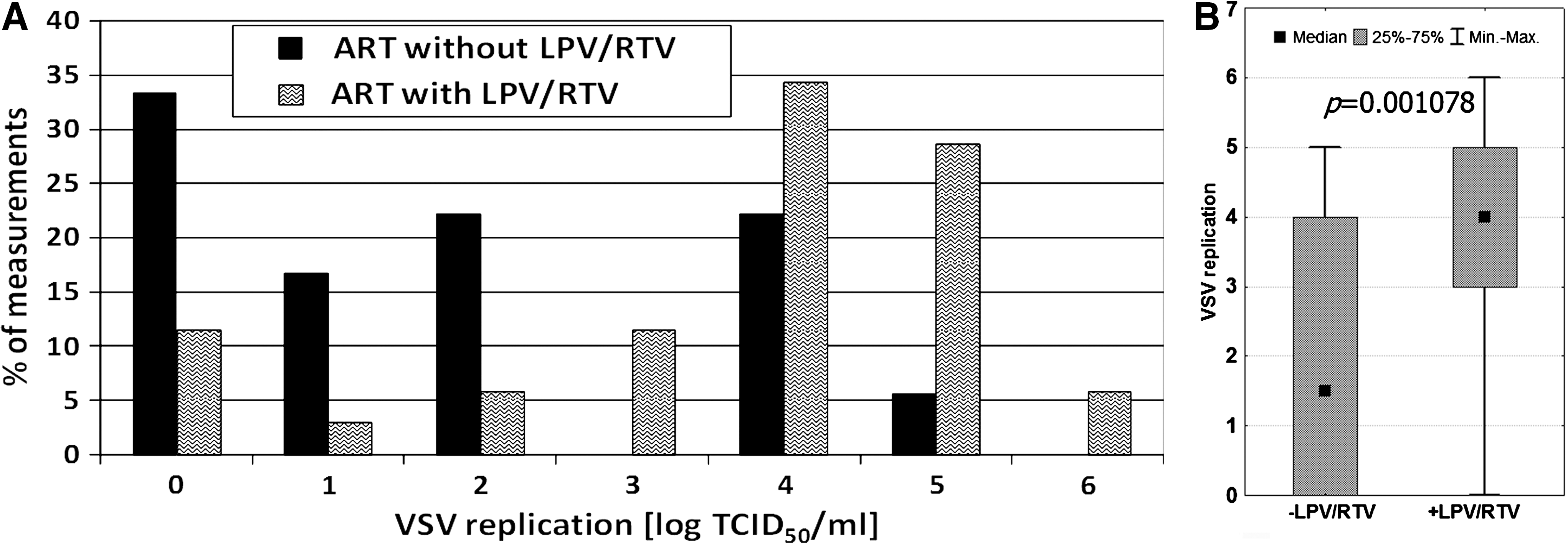
The effectiveness of antiretroviral therapy can depend on the combination of antiviral compounds included in the treatment regimen. Two groups of patients using ART were compared: (1) backbone therapy and LPV/RTV, and (2) backbone therapy and another protease or non-nucleoside inhibitor. The therapeutic scheme with LPV/RTV was chosen because clinical improvement was often observed in patients on such therapy. It was found that the replication of VSV was higher in the leukocytes of the LPV/RTV patients (Fig. 5). The mean log TCID50 VSV/mL for the LPV/RTV patients was 3.50, and for those without such treatment was only 2.04. The median values were 4 and 2, respectively. The results were statistically significant, suggesting a difference in clinical effect between patients on LPV/RTV therapy and those without it.
The differences in VSV replication level in the HIV-positive samples suggested that under the experimental conditions, both direct and indirect interactions between HIV and VSV could occur. Moreover, the effects could also result from direct drug interaction with the virus used. To test the latter possibility, LPV and RTV were chosen, since these drugs acted strongly, and their importance in therapy was significant. It was found that neither viricidal activity (Fig. 6), nor inhibition of VSV replication (data not shown), was directly caused by lopinavir or ritonavir at concentrations up to 1 mg/mL. Considering that the serum concentration of LPV or RTV in the blood was below 20 μg/mL (7,9), the results of our VSV determinations were not affected by the presence of these compounds in the blood samples. Moreover, the actual drug concentrations in the isolated cell cultures was significantly decreased due to the isolation procedure.
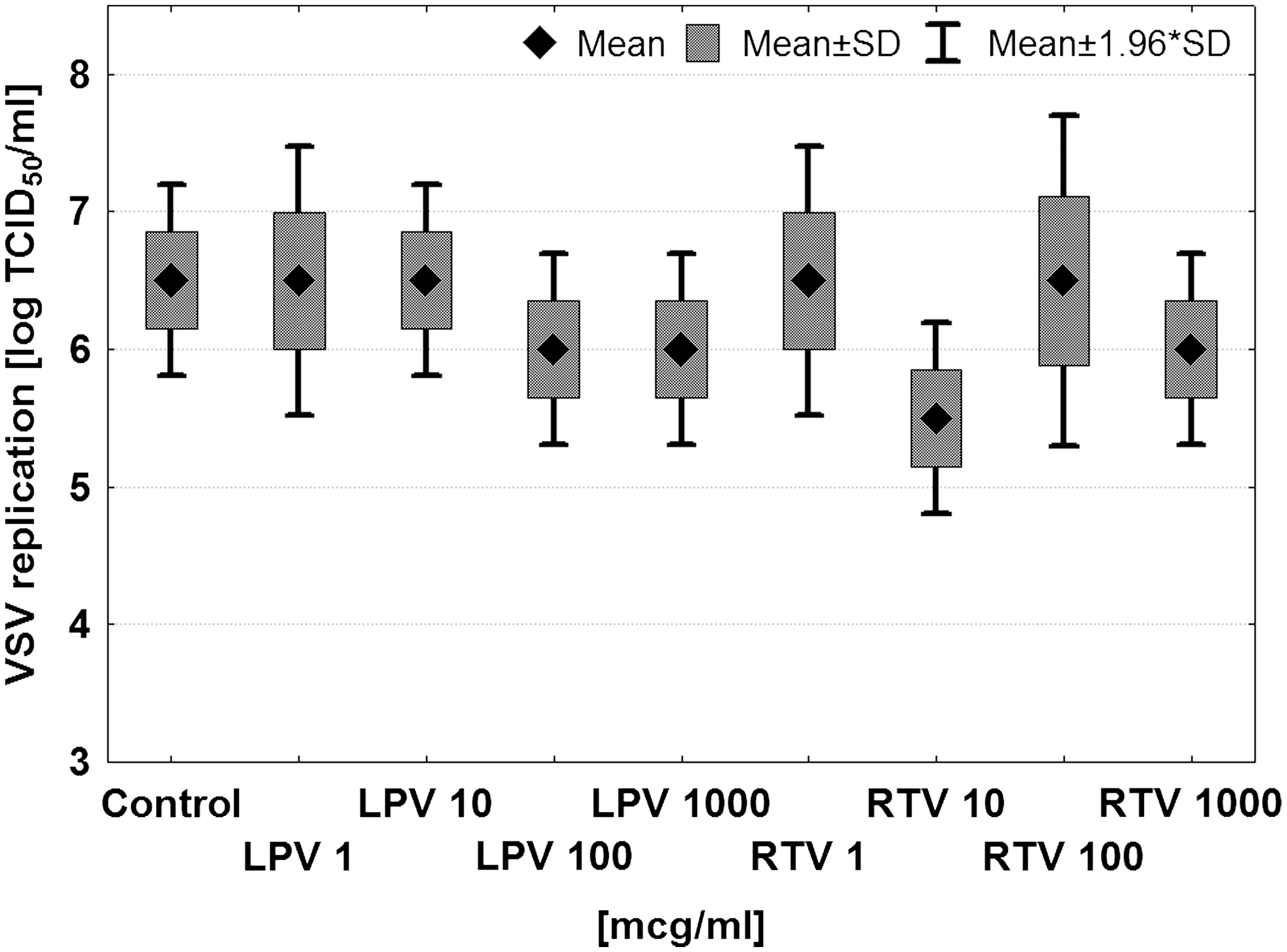
Effect of VSV treatment in vitro with lopinavir (LPV) or ritonavir (RTV). After direct VSV incubation with the given concentrations of the drugs, the viral titer was determined, showing no significant differences.
To check the viability of the isolated leukocytes, interferon induction with PHA was performed in all samples. In rare cases a lack of interferon production may be due to severe functional abnormalities of cells (suggesting a poor clinical status of the patient). In many cases it arises from technical errors in cell preparation before the assay is performed. Since interferon production was detected in all cases (12–288 IFN U/mL), the assay conditions were reliable.
In advanced HIV infection, detectable amounts of interferon could be found in plasma/serum. Up to several hundred units per milliliter could be detected in some cases (11,17,18). With regard to the antiviral activity of interferon, we determined whether the presence of this cytokine would affect VSV replication. Admittedly, the plasma had been washed out during the cell isolation procedure; however, the effects of interferon's action would still have been seen for several hours. There were 19 interferon-positive plasma samples (3–72 IFN U/mL) among those studied. The anti-VSV effect of interferon was actually observed, showing a significant reduction in VSV replication by leukocytes from patients with detectable (by biological assay, detection limit: 3 U/mL) amounts of the cytokine (Fig. 7). Biological assays based on antiviral activity are capable of measuring total interferon activity regardless of its type. Moreover, as type I (IFN-α and IFN-β) and type II (IFN-γ) interferons act synergistically, the biological IFN units better indicate the true power of its antiviral effect than enzyme immunoassays.
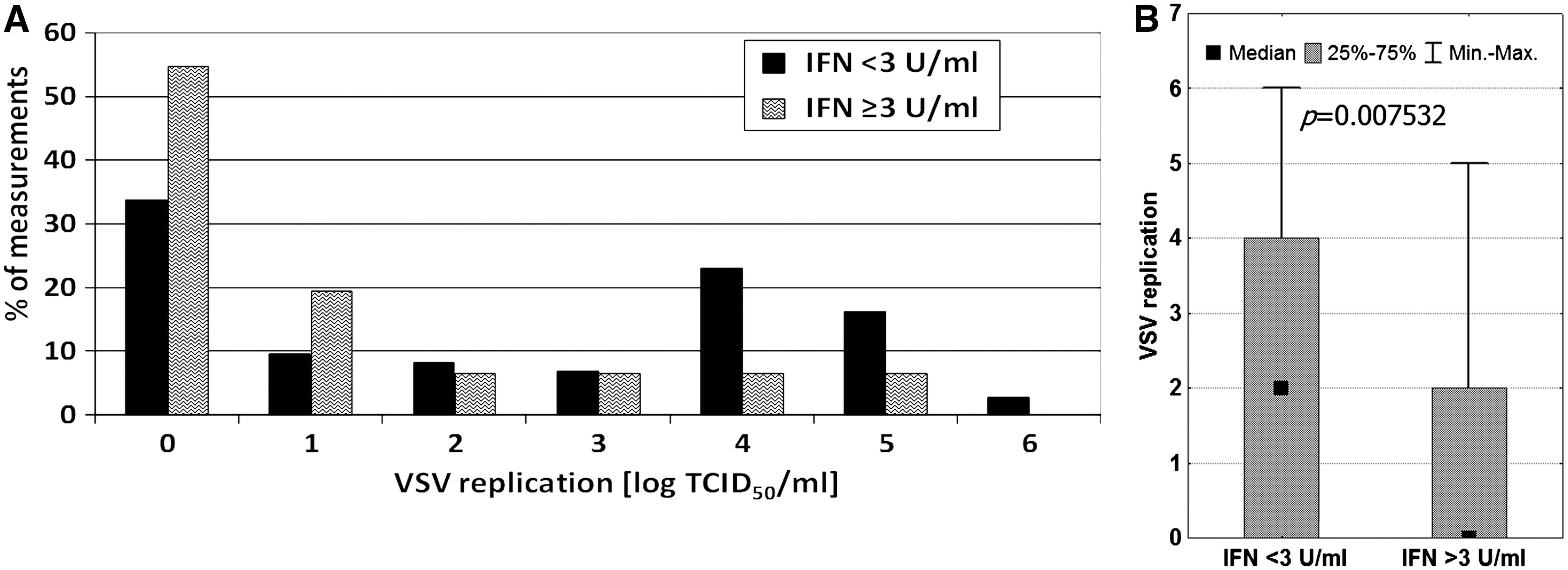
The results suggest that the presence of interferon could explain the inhibition of VSV replication seen in the cultures of leukocytes from HIV-infected patients. To check this hypothesis, the effects of viral load and antiretroviral therapy on VSV replication were reanalyzed after excluding the IFN-positive samples (Figs. 8 –10). No significant changes were observed in any case. However, in comparison with the previous analyses including all samples (Figs. 3 –5), the p values were found to be slightly higher (0.001579 versus 0.000089 for viral load analysis, 0.000018 versus 0.000001 for effect of therapy, and 0.001078 versus 0.001071 for the effect of LPV/RTV therapy), suggesting that endogenous interferon is only partially responsible for the inhibition of VSV replication.
Discussion
Measurement of the replication level of vesicular stomatitis virus (VSV) was used to determine the status of nonspecific antiviral immunity. Inhibition or a very low level of VSV replication was observed in healthy individuals. Along with the health deterioration caused by different diseases, the immune control of VSV infection becomes weaker, resulting in high levels of VSV titers (3,4,15,19,21,25).
It was to be determined whether similar effects could be found in HIV infection. The effects of immune system dysfunction are generally associated with the uncontrolled burst of miscellaneous pathogens. Therefore our initial assumption was that VSV replication should be poorly controlled in the leukocytes of advanced HIV-positive patients (high HIV viremia and low CD4+ cell count). This should result in VSV levels that were higher than in symptomless HIV-positive or healthy individuals. However, this hypothesis was not confirmed, as the results were quite the opposite, and suggested that the defense against VSV was not affected. Moreover, our observations strongly suggest that the participation of nonspecific mechanisms is able to inhibit VSV replication in leukocytes from patients with high HIV viremia.
To eliminate any doubts that potential VSV infection resulting in the development of specific anti-VSV immunity could interfere with the measurements on the VSV replication assay, prior to the assay patients potentially exposed to the virus were excluded from the study. In the other cases anti-VSV activity in the plasma was estimated. Since all tests performed were negative, we concluded that all subjects had no previous contact with the virus.
Generally, the profile of VSV replication was found to be similar in HIV-positive and HIV-negative persons (Fig. 1). However, the two peaks that we observed in the HIV-positive patients showed this population to be heterogeneous. This observation was not surprising. Along with the progression of the infection, deterioration in health leads to immune deficiency characterized by decreasing CD4+ cell counts, and the enhancement of viral load associated with it. Moreover, applying ART could reverse disease symptoms. The question was which progression marker primarily correlated with the inhibition of VSV replication. Analysis of the data indicated that viral load was the main factor (Fig. 3), with a stronger impact than CD4+ (Fig. 2) and CD8+ cell counts. This suggested that the presence of HIV and/or its replication in leukocytes acted on VSV replication. However, these two viruses belong to different families with different receptors, polymerases, and proteases. Thus there is likely no simple and direct interference with the replication cycle of VSV.
There are only a few reports on interactions between HIV and VSV. Kornbluth et al. (12) described complete inhibition of VSV replication in HIV-infected macrophages. Only monocytotropic HIV strains elicited such an effect, and it was mediated more by Mx protein than by interferons (1,12). On the other hand, Paik et al. (16) observed inhibition of HIV-1 and HIV-2 proviral expression by VSV matrix (M) protein. This protein was found to strongly inhibit host transcription (8). HIV-1 was successfully pseudotyped with VSV envelope glycoprotein (G) (2,13,14), and in the other direction the VSV G gene was replaced by the HIV env gene (5). In both cases, the cell specificity of the viruses was determined by the type of envelope protein. These experiments, though useful in vaccine and transduction studies, are hardly helpful in clarifying our ex vivo observations.
It is also possible that VSV replication was affected by the antiretroviral therapy. Actually, a significant effect of ART on VSV titer was observed (Fig. 4). Since particular therapeutic regimens might have different clinical efficacies, samples from patients currently on highly efficient therapy were chosen for separate analysis. The clinical examination showed that this assumption was fulfilled when lopinavir or ritonavir was included. The results (Fig. 5) showed that the application of LPV/RTV had a potent effect on VSV replication in leukocytes, similarly to that exerted in the persons with undetectable HIV viremia (Fig. 3). Since no direct viricidal effect of LPV and RTV on VSV was found (Fig. 6), the simplest explanation was that the observed differences in VSV replication were connected with HIV interference.
Viral interference is often mediated by interferons. Since the interferon system is impaired in advanced HIV infection (6,11,17,18,22), and its antiviral action is nonspecific, the effect of the cytokine on VSV has to be considered. In fact, detectable levels of interferon in plasma were accompanied by low levels of VSV replication (Fig. 7). However, the data analysis adjusted by exclusion of interferon-positive samples revealed no substantial changes (Figs. 8 –10). These findings are in agreement with earlier reports showing a lack of effect of interferon on the inhibition of VSV replication (1,12).
The results suggest that the inhibition of VSV replication was essentially caused by HIV. Regardless of whether a direct or indirect mechanism was engaged, the VSV replication assay was found to be useful in characterizing HIV-infected patients. In healthy persons, a high level of VSV replication reflects a weakening of innate immune mechanisms. The presence of some viral infections, including HIV, lead to interference with the sensitivity to VSV, resulting in the need to re-interpret the assay. In HIV infection the VSV replication level was found to be inversely correlated with HIV viral load. Therefore, it can be concluded that: (1) in most HIV-negative individuals VSV replication reflects the antiviral activity of innate immunity; (2) similarly, in early stages of symptomless HIV infection or after successful therapy (undetectable HIV viremia), the VSV replication level may generally result from the level of antiviral immunity; (3) in the case of viremic HIV patients, the level of VSV replication shows an inverse correlation with HIV viral load, and is therefore a useful indicator of current HIV activity; and (4) the VSV replication assay is also useful for monitoring antiretroviral therapy, and represents an important alternative to the standard determination of viral load and CD4+ cell count in HIV patients.
The measurement of VSV replication for monitoring HIV infection may be useful in cases when ambiguous results are obtained using standard assays. Regardless of the direct or indirect nature of the effects, the assay may be applied to evaluate the stage of HIV infection. Moreover, the method applied to other (not only viral) types of infections may have important prognostic value, in view of the fact that the monitoring methods used may be less well developed than those used to assess HIV infection.
Footnotes
Acknowledgments
This work was supported by grant no. 2 P05B 152 29 from the Ministry of Science and Higher Education, and grant no. ACA-POLA-02-1 from Abbott Laboratories.
Author Disclosure Statement
The work was partially supported by a grant from Abbott Laboratories, manufacturer of Kaletra. No other competing financial interests exist.