
Abstract
CD8+ T-cell-mediated pulmonary immunopathology in respiratory virus infection is mediated in large part by antigen-specific TNF-α expression by antiviral effector T cells, which results in epithelial chemokine expression and inflammatory infiltration of the lung. To further define the signaling events leading to lung epithelial chemokine production in response to CD8+ T-cell antigen recognition, we expressed the adenoviral 14.7K protein, a putative inhibitor of TNF-α signaling, in the distal lung epithelium, and analyzed the functional consequences. Distal airway epithelial expression of 14.7K resulted in a significant reduction in lung injury resulting from severe influenza pneumonia. In vitro analysis demonstrated a significant reduction in the expression of an important mediator of injury, CCL2, in response to CD8+ T-cell recognition, or to TNF-α. The inhibitory effect of 14.7K on CCL2 expression resulted from attenuation of NF-κB activity, which was independent of Iκ-Bα degradation or nuclear translocation of the p65 subunit. Furthermore, epithelial 14.7K expression inhibited serine phosphorylation of Akt, GSK-3β, and the p65 subunit of NF-κB, as well as recruitment of NF-κB for DNA binding in vivo. These results provide insight into the mechanism of 14.7K inhibition of NF-κB activity, as well as further elucidate the mechanisms involved in the induction of T-cell-mediated immunopathology in respiratory virus infection.
Introduction
The adenovirus genome E3 region contains several genes that encode proteins which inhibit TNF-α- or Fas L-mediated apoptosis of target cells (4,22), and the 14.7K protein has been shown to protect against in vitro cytolysis by CD8+ T cells or by TNF-α (13). Compared with the wild-type adenovirus, mutants lacking 14.7K resulted in enhanced inflammatory influx and lung injury in murine adenovirus pneumonia (38). Transgenic mice expressing 14.7K in the alveolar epithelium are resistant to LPS-mediated lung inflammation, and displayed a decrease in mononuclear influx (16). However, signal transduction events involved in adenoviral 14.7K regulation of gene epithelial expression are poorly understood, notwithstanding the fact that analysis of these pathways is likely to inform the mechanisms of immunopathology associated with a variety of respiratory epithelial infections. In this report, we investigated the effects of epithelial adenoviral 14.7K expression on CD8+ T-cell- and TNF-α-mediated NF-κB activation, CCL2 production, and the induction of pulmonary immunopathology in influenza infection.
Materials and Methods
BALB/c, SP-C-14.7K, and SP-C-HA-14.7 mice were used at 12–16 wk of age. The SP-C-14.7K and SP-C-HA mice have been backcrossed onto the BALB/c background for 10 generations. The double transgenic (SP-C-HA-14.7) strain was generated by interbreeding mice expressing either HA or 14.7 under the transcriptional control of the surfactant protein C (SP-C) promoter, which directs expression to the distal airway (alveolar and bronchiolar) epithelium (10,16). All experiments were conducted in strict accordance with the guidelines of the National Institute of Health (NIH) and the Dartmouth Medical School Institutional Animal Care and Use Committee (IACUC), including the requirement that any animal that lost 20% of its initial weight after infection would be euthanized. CD8+ T-cell bulk lines and clones were activated in vitro prior to adoptive transfer (43), and injected via the tail vein. Weight loss was monitored daily, and lungs were harvested at appropriate times for histology or chemokine analysis. Lung tissue extracts were assayed for CCL2 using a sandwich ELISA (BD Pharmingen, San Diego, CA), according to the manufacturer's instructions. In some experiments, mice were infected intranasally with 5 × 103 TCID50 of influenza A/PR/8/34. Five days after virus infection, the animals were anesthetized, a tracheostomy tube was inserted, and whole-lung diffusing capacity was determined by CO uptake (a surrogate for oxygen transfer), as previously described (11). In some experiments the partial arterial pressure of oxygen was measured in samples from the ventral tail artery, as previously described (42). Viral titers in whole-lung homogenates were determined as TCID50 in MDCK cells (43). Mouse TNF-α and IFN-γ were purchased from Genzyme (Boston, MA) and PBL (New Brunswick, NJ), respectively. For in vitro studies, murine alveolar epithelial cells stably expressing H-2Kd (MLE-Kd; 46) were treated with synthetic peptide representing the Kd-restricted 210–219 epitope of the A/Japan/57 HA and co-cultured with HA210-specific CD8+ T cells, or treated with soluble TNF-α (20 ng/mL) or IFN-γ (1000 U/mL). In co-culture experiments, MLE-Kd and CD8+ T cells were separated after incubation using mouse anti-CD8-bound magnetic beads. Total RNA was prepared from cells using the RNeasy kit (Qiagen, Valencia, CA). RT-PCR was performed using primer sequences as previously described (32), and RETROscript (Ambion, Austin, TX), according to the manufacturer's protocol. Cell extracts were prepared and proteins were separated by electrophoresis using 8–10% SDS-PAGE gels (25). Proteins in the gel were electrophoretically transferred to polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA), subjected to immunoblotting with the indicated antibodies, and visualized by enhanced chemiluminescence (Pierce Protein Research Products, Rockford, IL). Antibodies to IκB-α and NF-κB (p50 and p65) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies to phospho-Akt (serine 473), Akt, phospho-GSK-3β, GSK-3β, phospho-p65 (S468), and horseradish peroxidase (HRP)-linked secondary antibodies were purchased from Cell Signaling Technology (Boston, MA). Chromatin immunoprecipitation (ChIP) assays were performed using the EZ-ChIP kit (Upstate/Millipore, Billerica, MA), according to the manufacturer's directions, using PCR primers spanning the NF-κB site in the CCL2 promoter (39).
Results
To study events in intraepithelial TNF-α signaling that lead to inflammatory chemokine induction upon CD8+ T-cell recognition, we stably expressed the adenoviral 14.7K protein in MLE-Kd alveolar epithelial cells, and studied CCL2 induction triggered either by T-cell antigen recognition, or by treatment with soluble TNF-α. As shown in Fig. 1A, both CD8+ T-cell recognition and treatment with recombinant TNF-α induced CCL2 expression in control MLE-Kd cells, but this was significantly abrogated in 14.7K-expressing MLE-Kd. The effect of 14.7 appeared to be specific for CCL2, because induction of another important chemokine, CXCL2 (MIP-2), was not significantly altered under similar conditions. In addition, 14.7K expression had no impact on the expression of guanylate binding protein (GBP-2) and IFI-47, genes known to be induced by interferon-γ, another antigen-dependent product of CD8+ T cells.

Stable expression of adenovirus 14.7K abrogates NF-kB activation and CCL2 induction by CD8+ T-cell recognition or TNF-α treatment in MLE-Kd cells.
The NF-κB pathway plays an important role in TNF-α induction of CCL2 expression (29). The predominant NF-κB heterodimer consists of p65 and p50 subunits in the cytoplasm, and are bound to an inhibitor of NF-κB (Iκ-Bα) protein that prevents its activation and translocation into the nucleus (12,28). We therefore tested whether the adenoviral 14.7K protein would inhibits Iκ-Bα degradation or p65 nuclear translocation. Western blot analysis revealed that adenoviral 14.7K did not inhibit TNF-α-induced Iκ-Bα degradation or p65 nuclear translocation in MLE-Kd cells (Fig. 1B). Recruitment of NF-κB subunit p65 to the NF-κB-responsive element has been shown to be a major regulatory event in transcriptional activation by TNF-α (29,39). ChIP revealed that the p65 and p50 subunits were recruited to the CCL2 promoter in response to TNF-α in control MLE-Kd but not in adenoviral 14.7K-expressing cells (Fig. 1C), suggesting that adenoviral 14.7K inhibits transcriptional activation of CCL2 by interfering with NF-κB recruitment to its promoter, rather than by inhibition of nuclear translocation. In order to understand the mechanisms involved in this inhibition, we examined several other candidate signal transduction pathways triggered by TNF-α signaling. In addition to the IKK pathway, TNF-receptor activation regulates multiple pathways in parallel, including mitogen-activated protein kinases (MAPK), such as extracellular-regulated kinases (ERK), Jun N-terminal kinases (JNK), and p38 kinase. In addition, TNF-α signaling may regulate phosphoinositide 3′-kinase (PI3K), Akt, and glycogen synthase kinase-3β (GSK-3β), which are involved in multiple phosphorylation events and the recruitment of various transcription factors and co-regulators (12,28,39,40,44). Among these kinases, GSK-3β is unique in that it is constitutively active under normal conditions and inactivated in response to PI3K/Akt-mediated inhibitory phosphorylation (9). Interestingly, GSK-3β has been reported to be required for NF-κB recruitment to κβ-responsive elements, and induction of a subset of NF-κB regulated genes, including CCL2, but not CXCL2 (39). TNF-α has been reported to stimulate phosphorylation on serine9 of GSK-3β in a variety of cells (14,15,19), so we performed Western blot analysis on 14.7K-expressing MLE-Kd cells for GSK-3β after treatment with TNF-α. As shown in Fig. 1D, TNF induction of serine9 phosphorylation of GSK-3β was attenuated in MLE-Kd cells expressing 14.7K compared with control MLE-Kd cells, suggesting that this may be an important mechanism regulating at least some of the transcriptional activities induced in epithelial cells by TNF-receptor signaling. In an effort to further understand the mechanisms of this inhibition, we performed a similar analysis of serine473 phosphorylation of Akt, which is upstream of GSK-3β, and found that this was also attenuated by 14.7K (Fig. 1D). In contrast, TNF-α-induced ERK and p38 phosphorylation was unaffected in both cells (data not shown). TNF-α-induced serine9 phosphorylation of GSK-3β has been shown in other studies to be important for the serine468 phosphorylation of the p65 subunit of NF-κB (15), and therefore we asked whether 14.7K expression would impact TNF-α induction of serine468 phosphorylation of p65 in MLE-Kd cells. Western blot analysis revealed that serine468 phosphorylation of p65 was indeed attenuated in 14.7K-expressing MLE-Kd cells compared with control cells (Fig. 1D). Abrogation of serine phosphorylation of GSK-3β and p65 by adenoviral 14.7K may be a key mechanism by which TNF-α-receptor induction of NF-κB activation and CCL2 expression are suppressed in MLE-Kd cells, and suggests that this may represent an important regulatory event in epithelial antiviral immunity.
To evaluate the physiological and pathological consequences of inhibiting this aspect of epithelial TNF-receptor signaling in the setting of an effector CD8+ T cell response to an acute virus infection, we used a well-characterized model involving intranasal influenza infection, followed by adoptive transfer of HA-specific CD8+ effector cells (45). Transgenic animals expressing the 14.7K protein on the distal airway (alveolar and bronchiolar) epithelium under control of the surfactant protein C promoter (16) were infected and then used as recipients of activated CD8+ T cells transferred intravenously. As shown in Fig. 2A, infection of both transgenic and WT animals with A/PR8/34 resulted in a considerable decrement in CO uptake, a surrogate measure for oxygen transfer capability (or diffusing capacity; 11), and this was significantly worse in WT mice after transfer of CD8+ effectors, as we have previously shown (46). Interestingly, there was no additional decrement in the 14.7K transgenic mice after T-cell transfer compared to infection alone, suggesting that the injury attributable to CD8+ T-cell effector activities was ablated by inhibition of this epithelial TNF-receptor-dependent signaling pathway by 14.7K. CD8+ T-cell transfer alone (without infection) had no impact on CO uptake, and these measurements were similar to those observed in unmanipulated mice (data not shown). Clearance of virus was delayed in 14.7K transgenic mice as well, which had detectable lung viral titers 4 d after infection and transfer, while the WT animals did not (Fig. 2B).
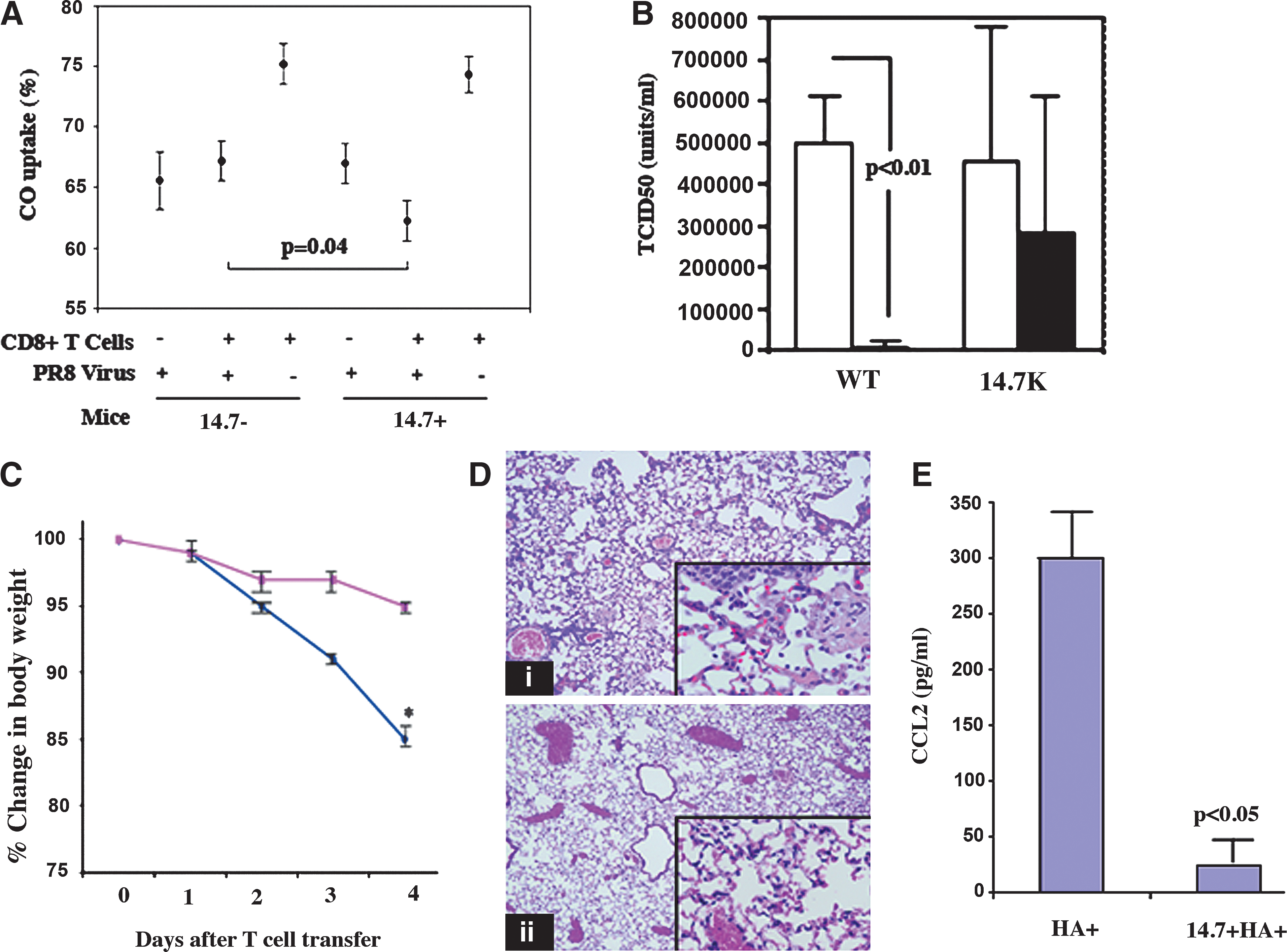
Adenoviral 14.7K expressed in alveolar epithelium is protective against CD8+ T-cell-mediated lung injury in vivo.
In order to specifically focus on the immunopathology triggered by antigen-specific T-cell activities, without the variable of viral clearance, we performed a set of experiments in a model in which lung injury is mediated exclusively by CD8+ T-cell recognition of transgenically-expressed target antigen in the distal airways. Mice expressing both 14.7K and influenza HA on the alveolar epithelium were generated by breeding 14.7K transgenic mice with mice expressing HA under the control of the same promoter (10), resulting in alveolar epithelial expression of both transgenes. Adoptive transfer of activated HA-specific CD8+ T cells IV resulted in significant morbidity (as measured by weight loss) in WT-HA+ compared with 14.7K-HA+ mice (Fig. 2C). All of the WT HA+ animals were euthanized after day 4 because of their weight loss. Pulmonary infiltrates in WT-HA+ lungs consisted largely of a macrophage and neutrophil influx, whereas the macrophage influx was attenuated in 14.7K-HA+ mice (Fig. 2D). Consistent with the histopathology, CCL2 expression in lung was significantly attenuated in 14.7K-HA+ compared with WT-HA+ mice (Fig. 2E). Measurement of arterial P
Discussion
The TNF-α and NF-κB pathways are central regulators of immune responses, cell survival, and apoptosis, and are modulated by a large number of pathogens for their survival (31,33). There are numerous examples of evasive strategies that unrelated viruses have evolved to counter these systems (2,27), in addition to adenovirus, indicating their potential significance in antiviral immune responses. In this study, we focused primarily on the immunopathological impact of inhibition of this pathway by 14.7K, using influenza infection as a model in which immune-mediated lung injury is likely an important contributor to the clinical outcome. We have previously shown that CD8+ T-cell expression of TNF-α and alveolar epithelial expression of TNF-R1 and TNF-R2 on lung epithelial cells are required for significant T-cell-mediated lung injury (24,43), and that induction of epithelial chemokine expression, particularly CCL2, is a primary contributor to the immunopathology (37,45). In this study, we found that distal airway epithelial CCL2 induction was abrogated by adenoviral 14.7K through the inhibition of TNF-α-receptor-induced GSK-3β phosphorylation and recruitment of NF-κB to the CCL2 promoter. GSK-3β is required for NF-κB recruitment and induction of a subset of NF-κB-regulated genes including CCL2, but not CXCL2 (39). Overexpression of GSK-3β has been shown to enhance, while its inhibition has been shown to abrogate, CCL2 induction and the inflammatory responses associated with chronic renal allograft disease (14). Targeted deletion of GSK-3β in mice has been shown to abrogate NF-κB activation mediated by TNF-α (19). Whether the 14.7K protein inhibits Akt and GSK-3β serine phosphorylation directly or by modulation of upstream signaling pathways remains to be determined. In any case it provides an interesting tool for analyzing the critical biochemical underpinnings of the immunopathological responses to virus infection, and of those mediated primarily by the host immune response.
Our in vivo experiments support previous findings indicating the importance of TNF-α in CD8+ T-cell-mediated lung injury (43,46). Although the system we use arguably reflects a model of secondary rather than primary T-cell responses to influenza infection, it provides a unique opportunity to focus our studies on the epithelial responses to activated T-cell recognition in viral infection, which occur much more slowly in the primary response. During influenza infection, viral antigens are presented on both epithelial and non-epithelial cells in the lungs, yet interference with TNF-α signaling exclusively in distal airway epithelial cells resulted in abrogation of CD8+ T-cell-mediated lung injury (as well as viral clearance). This underscores the importance in influenza infection (and undoubtedly others) of the interaction between CD8+ T cells and airway epithelial cells as the key event in immune-mediated lung inflammation and viral clearance. Understanding the signal transduction pathways involved in host epithelial inflammatory responses and viral evasion/inhibition of these pathways may lead to novel strategies to mitigate pulmonary immunopathology in respiratory virus infection.
Footnotes
Acknowledgments
This work was supported by grants AI083024, AI069360, AI45221, and RR16437 from the National Institutes of Health.
Author Disclosure Statement
No conflicting financial interests exist.