
Abstract
Chemokines play a pivotal role in the innate response to both bacterial and viral infections, and in mixed infections. To determine chemokine responses to Sindbis virus (SIN) in a co-infection model, peripheral blood mononuclear cells (PBMCs) derived from healthy volunteers were exposed to SIN in the presence and absence of lipopolysaccharide (LPS). Culture supernatants recovered at 2, 24, and 72 h post-exposure were evaluated for virus replication and analyzed for chemokines by ELISA. None of the PBMC cultures showed new virus release, GFP reporter expression, or viral RNA synthesis. While SIN had little effect on the induction of IL-8 and RANTES, the chemokines MCP-1, MIP1-α (p < 0.001), and MIP1-β (p < 0.0004) were drastically upregulated by SIN as well as LPS. Both live and UV-inactivated SIN induced secretion of IP-10 and I-TAC. Although LPS did not induce release of IP-10, it sharply inhibited (p = 0.004) SIN-mediated IP-10 secretion. On the contrary, the release of SLC was blocked by SIN. The adjuvant activity of IP-10, its antiangiogenic function, and antagonism between SIN and LPS for the release of select chemokines may be useful in understanding the pathogenesis of mixed infections, cross-talk between cellular pathways, and may have applications in cancer and sepsis.
Introduction
Sindbis virus is one of the best studied mosquito-transmitted RNA viruses (34). Occasionally, it causes fever and arthralgia in humans that is much milder than Chikungunya arthritis (13). Genetically-engineered RNA genomes of alphaviruses including Sindbis virus (SIN), Semliki Forest Virus (SFV), and Venezuelan Equine Encephalitis Virus (VEE) are extensively used as viral vectors (8,28), and as oncolytic agents (37). Using mouse models, the inflammatory response including type I interferons, TNF-α, and IFN-γ to alphaviruses have been well studied (12,32). Strikingly, chemokines appear to contribute to alphavirus pathogenesis and arthralgia (31). In the present work, we examined the chemokine response of human peripheral blood mononuclear cells (PBMCs) to SIN exposure. The bacterial component, lipopolysaccharide (LPS), is one of the best studied TLR-4 ligands in inflammation research (2,14). LPS downregulates interferon induction by blocking virus-induced phosphorylation of IRF-3 and IRF-7 (15). We hypothesized that LPS might alter SIN-induced chemokine release and vice versa, and this knowledge may shed light on the pathogenesis of mixed infections.
Chemokines are a family of small secreted proteins that mediate inflammation, angiogenesis, and lymphocyte trafficking (10). They are grouped under four subfamilies: C, CC, CXC, and CX3C, based on the location of N-terminal cysteine residues. Chemokine receptors are a multitude of G-protein coupled receptors. Each type of receptor can bind to several chemokines, and each chemokine can bind to more than one receptor. Both bacterial and viral infections induce common and distinct chemokines (10). The CXC chemokine IP-10 is induced by type I and type II interferons, and is secreted from monocytes and other cell types (11,21). In addition to bacterial and viral infections, IP-10 is expressed at high levels in autoimmune disorders (10,38). Since alphaviruses are an important cause of arthralgia, the study of virus-induced chemokine expression may be relevant (31). However, the regulation of IP-10 induction during mixed infections and how it modifies disease outcome is unclear.
Robust SIN replication occurs in a variety of vertebrate cell types and insect cells in culture (28,34). Although human PBMCs and lymphoid tissue are known to be refractory to SIN replication, the virus can be adapted to replicate in human dendritic cells (9,35). Human macrophages were recently shown to be infected with Sindbis strain MRE16 (1). Previous work of others indicated significant innate responses for some viruses in the absence of replication (33,39). In the present work, we show differential chemokine induction by SIN, and pinpoint antagonism for the release of select chemokines by LPS. Possible mechanisms and applications of these findings are also discussed. To our knowledge, this is the first report on SIN-mediated IP-10 induction in PBMCs in the absence of virus replication.
Materials and Methods
Cell culture
BHK and Vero cells were maintained in minimal essential medium (Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum. BHK cells were used for transfection of in vitro transcribed RNA, and Vero cells were used for production and titration of SIN Toto1101 stocks (29). Unless specified, all reagents were purchased from Invitrogen.
In vitro transcription of SIN expression plasmids
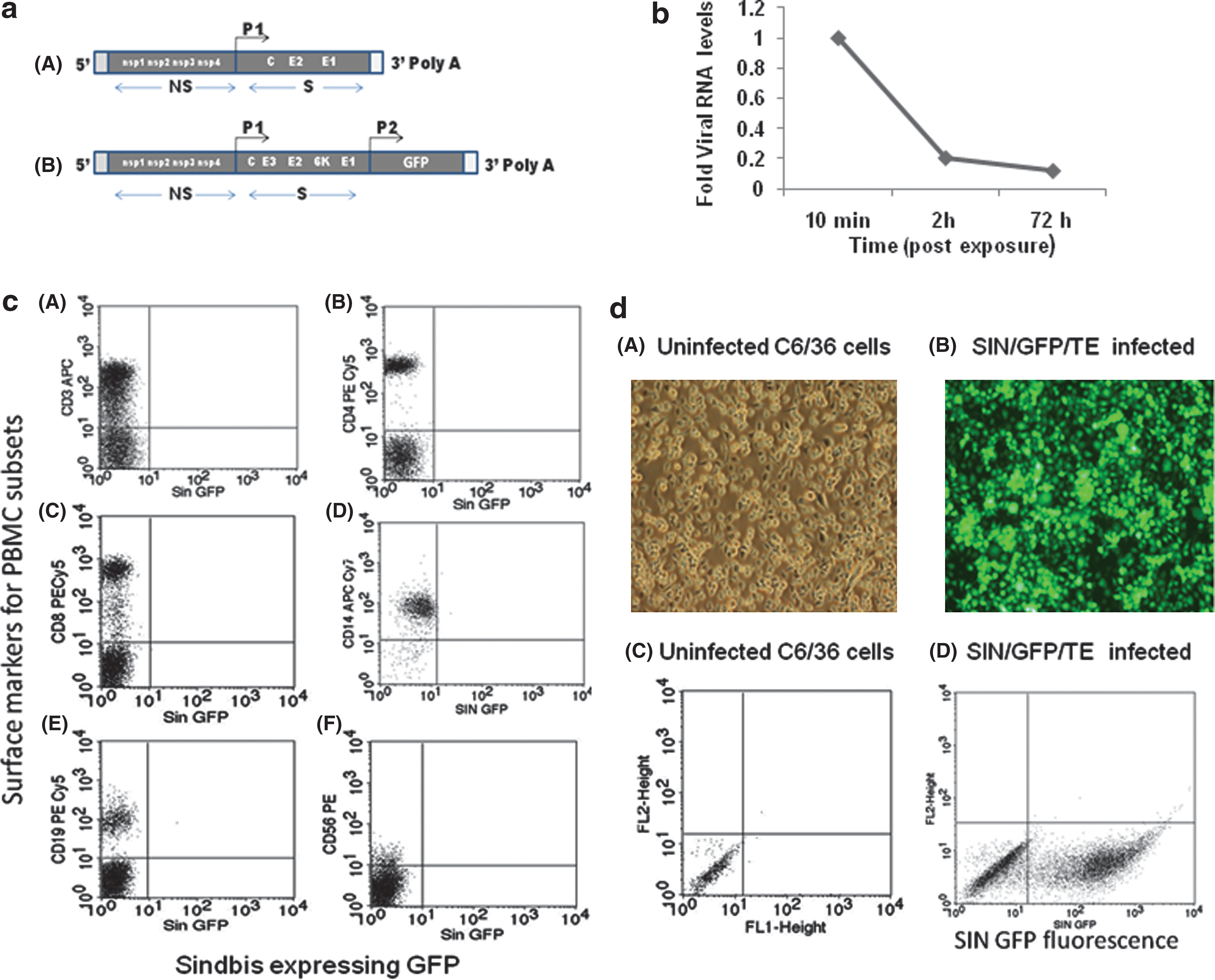
In vitro transcription of SIN Toto1101 and SIN/GFP/TE was done as described earlier (29) (Fig. 1). Approximately 4 μg SIN Toto1101 and SIN/GFP/TE (8) was linearized with Xho1 (New England Biolabs, Ipswich, MA) and subsequently ethanol precipitated and dissolved in DEPC-treated water. The linearized plasmids were then transcribed in vitro using SP6 RNA polymerase in 30 μL transcription mix containing 0.5 mM NTP, 10 mM DTT, and cap analogs. The transcription mixture was incubated at 37°C for 1 h. Subsequent to DNAse treatment and phenol-chloroform extraction, RNA was precipitated and washed with ethanol, dissolved in DEPC-treated water, and stored at −20°C.
(
RNA transfection and production, purification, and endotoxin testing of virus preparations
Confluent BHK cells in 100-mm tissue culture dishes were transfected with 250 μL of transfection mixture containing 3 μg of the transcribed RNA and 20 μL of Lipofectin (29). The plates were continually rocked for 20 min at room temperature. After 20 min, the transfection mixture was removed and 1 mL of prewarmed MEM containing 10% fetal bovine serum and gentamicin (50 μg/mL) was added to the cells and incubation was continued at 37°C. The cells were observed for cytopathic effect and cell-free culture supernatants recovered after 24 h were stored at −20°C. Stock SIN and SIN/GFP/TE viruses were prepared in Vero or BHK cells by infecting at a MOI of 1 and incubation at 37°C for 48 h. Virus seeds were prepared by clarifying infectious cell culture media by centrifugation and stored in aliquots at −80°C. The virus was further purified by ultracentrifugation of culture fluid for 2 h at 30 K rpm under a 20% sucrose cushion. Both sucrose-cushion purified and the parental virus preparations behaved similarly. UV-inactivation of virus particles was carried out in a Bio-Rad UV-chamber (program C4x2; Bio-Rad, Hercules, CA). Endotoxin testing of virus preparations was carried out using the Kinetic Chromogenic Assay Kit using LAL reagent (Charles River Laboratories, Wilmington, MA) per the manufacturer's protocol, and were found to be less than 0.005–0.0068EU/million pfu of virus or UV-inactivated virus equivalents. Viral titers were determined by plaque assay with serially diluted virus stock on Vero cell monolayers. No infectious virus was detectable even in undiluted UV-inactivated virus preparations.
Peripheral blood mononuclear cells
Healthy volunteers were recruited for the study after approval of the protocol by the Institutional Review Board (IRB) of Meharry Medical College, and peripheral blood was collected in heparinized tubes. PBMCs were separated by density gradient centrifugation, using Ficoll-Paque plus. After washing in HBSS, PBMCs were cultured in RPMI-1640 medium supplemented with 2 mM glutamax containing 5% autologous plasma at a concentration of 2 × 106 cells/mL in 12-well plates. Cell viability was checked using trypan blue dye exclusion testing, and more than 95% of the cells were viable.
In vitro induction of peripheral blood mononuclear cells
Fresh PBMCs collected from healthy subjects were stimulated with SIN or LPS (Imgenex, San Diego, CA) in vitro to induce chemokine production and secretion into the culture supernatant fluids. Briefly, PBMCs (2 × 106/mL) were treated with SIN/GFP/TE, SIN (Toto1101; MOI of 5), or/and E. coli LPS (1 μg/mL) and maintained for 72 h at 37°C and 5% CO2. Supernatants were collected from the cell cultures after 2, 24, and 72 h of incubation at 37°C in 5% CO2, clarified by centrifugation, and frozen at −80°C for later chemokine and viral plaque assays. SIN/GFP/TE-infected cells were removed with cold phosphate-buffered saline (PBS, pH 7.2), washed, and stained for surface markers, and analyzed by flow cytometry. In parallel, C6/36 mosquito cells were also infected with SIN/GFP/TE at MOI of 1 for 24 h at 30°C in 5% CO2 and observed under fluorescent microscopy.
Phenotyping of SIN GFP-infected PBMCs by flow cytometry
Surface staining of the PBMCs infected with SIN/GFP/TE was done to determine SIN-specific RNA synthesis and GFP expression at 2, 24, and 72 h using APC-conjugated anti-CD3 antibody, PE-Cy5-conjugated anti-CD4, CD8, and CD19 antibodies, and PE-conjugated anti-CD56 antibody. The monocytes were stained with anti-CD14-APC antibody (BD Biosciences Pharmingen, Franklin Lakes, NJ). The stained cells were washed in PBS, fixed with 1% PFA, and acquired in a FACSCalibur flow cytometer. By scatter pattern, 10,000 events were counted in the lymphocyte- and monocyte-gated regions and analyzed using CellQuest Pro software.
Chemokine assay in culture supernatants
The levels of CCL2/MCP-1, CCL3/MIP-1α, CCL4/MIP-1β, CCL5/RANTES, CCL21/SLC, CXCL8/IL-8, CXCL10/IP-10, and CXCL11/I-TAC in culture supernatants were determined using DuoSet ELISA Development Systems (R&D Systems, Minneapolis, MN), according to the manufacturer's instructions. The detection ranges for these assays were 15.6–1000 pg/mL for CCL5, CXCL11, CCL21, CCL2, CCL4, and 31.25–2000 pg/mL for CXCL8 and CXCL10, and 7.8–500 pg/mL for CCL3. To minimize interassay variability of measurements, samples of each study subject were assayed for respective chemokines in the same ELISA plate on the same day of assay.
Plaque assay of PBMC supernatants
To determine the viral titer, PBMC supernatants collected at 2, 24, and 72 h were serially diluted and used to infect Vero cell monolayers. After 1 h of infection, the viral suspension was removed and the monolayers were overlayed with 1% noble agar in MEM and incubated at 37°C. After 72 h, the cells were observed for the appearance of plaques, stained with crystal violet, and the plaques were counted (29).
Statistical analysis
Differences in mean cytokine levels were compared using paired Student's t-test and the non-parametric Wilcoxon signed ranks test wherever applicable using Microsoft Excel and GraphPad Prism 5.0. The results were expressed as arithmetic mean ± standard error. p Value <0.05 was set as the level of significance.
Results
Lack of SIN GFP expression and virus release in PBMCs
To determine SIN replication in PBMCs, we used infectious virus derived from an engineered expression plasmid, Toto1101 (Fig. 1a). PBMC culture supernatants exposed to SIN were analyzed by viral plaque assay. Except for the amount of input virus, additional virus release at 2, 24, and 72 h was not demonstrable (data not shown). Parallel PBMC cultures were exposed to an engineered SIN that encodes the GFP reporter protein SIN/GFP/TE (Fig. 1c), and the cells were analyzed by flow cytometry. No GFP fluorescence was observed for any of the PBMC populations tested (Fig. 1c), while a high level of GFP expression was observed in infected C6/36 cells (Fig. 1d). These results indicated defective RNA synthesis and virus release from PBMCs exposed to SIN in the presence or absence of LPS. Determination of virus-specific RNA from PBMCs by quantitative PCR also indicated a lack of accumulation of virus-specific RNA (Fig. 1b).
SIN has a minimal effect on CXCL8/IL-8 and CCL5/RANTES secretion
Very low IL-8 release was observed at 2 h in all culture conditions (Fig. 2A). However, at 24 and 72 h, LPS- as well as SIN + LPS-treated cultures showed high levels of IL-8. This result suggests that SIN did not antagonize LPS-induced IL-8 release. The level of RANTES was comparable at the 2- and 24-h time points for infected and uninfected cultures (Fig. 2B). However, at 72 h, LPS-treated, and SIN + LPS-treated cultures showed significantly higher levels (p = 0.001, p = 0.0007) of RANTES, suggesting that the LPS-induced RANTES level was unaltered by SIN co-exposure.
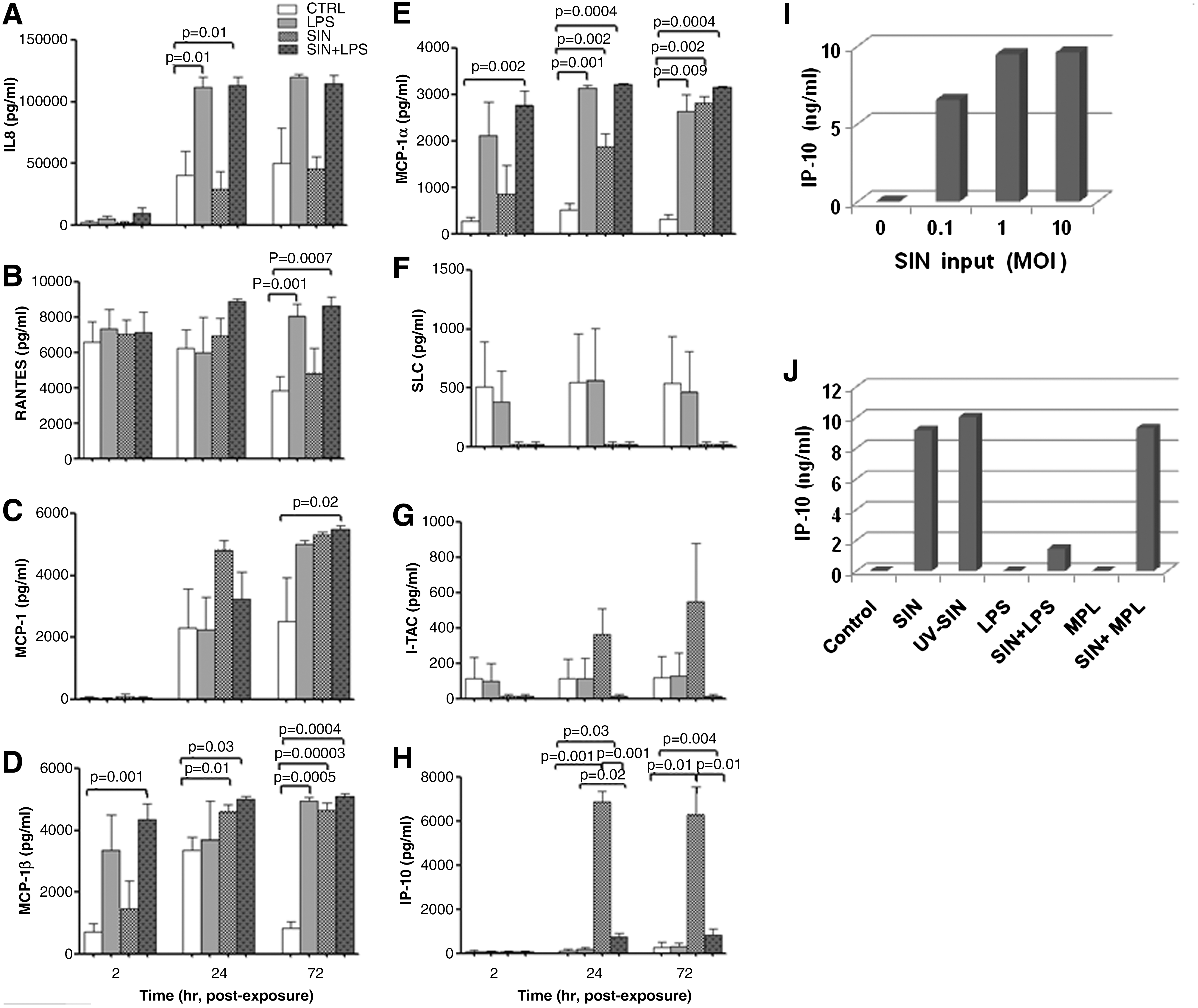
Chemokine secretion in culture supernatants of PBMCs from healthy subjects (n = 4) exposed to SIN (MOI 5), LPS (1 μg/mL), and SIN + LPS was estimated by ELISA at 2, 24, and 72 h. (
SIN as well as LPS stimulates CCL2/MCP-1, CCL3/MIP-1α, and CCL4/MIP-1β
At 2 h negligible amounts of MCP-1 were detectable in all the culture supernatants (Fig. 2C). However at 24 and 72 h, moderately high levels of MCP-1 were released from SIN, LPS, and SIN + LPS (p = 0.02) treated cultures. These data suggest non-antagonizing pathways for LPS and SIN in upregulating MCP-1 release. As shown in Fig. 2E, even at 2 h, high levels of MIP-1α were secreted in LPS- and SIN + LPS-treated (p = 0.002) cultures. At 24 and 72 h, all of the three treated cultures released high (p < 0.001) amounts of MIP-1α, whereas control cultures released very low levels of MIP-1α. These results suggest that there was a delay in SIN-induced secretion of MIP-1α, compared to LPS. Similarly high levels of MIP-1β (p < 0.0005) were found for all treated cultures at 72 h (Fig. 2D).
Antagonism between LPS and SIN for IP-10 and SLC induction
The chemokines IP-10 and I-TAC interact with the same receptor, CXCR3. As shown in Fig. 2H, very little IP-10 was detectable at 2 h in all cultures. However, at 24 and 72 h, significantly high levels (p = 0.001) of IP-10 were induced by SIN. Although LPS did not induce any IP-10, it drastically (p = 0.001) blocked the SIN-induced IP-10 release. Interestingly, I-TAC induction by SIN was also blocked by LPS (Fig. 2G), suggesting the involvement of a common pathway. At all time points tested, control and LPS-treated cultures expressed moderate amounts of SLC (Fig. 2F). However, SIN effectively blocked the release of SLC, both in the presence and the absence of LPS. Table 1 summarizes the chemokines that were analyzed, and how LPS and SIN influenced their secretion in a parallel/antagonistic manner.
Upward or downward arrows denote an increase or decrease in chemokine levels, while ‘–’ indicates null effect compared to untreated PBMCs.
As shown in Fig. 2I, the level of IP-10 induction was high even at 0.1 MOI of SIN exposure, and only a twofold further increase could be achieved at the MOI of 1 and 10. Interestingly, even UV-inactivated SIN induced comparable levels of IP-10, further indicating that virus replication in PBMCs is not required for IP-10 induction (Fig. 2J). Remarkably, monophosphoryl lipid A, MPL, a LPS derivative used as a vaccine adjuvant (30), did not inhibit virus-induced IP-10 secretion (Fig. 2J). This observation suggests that other chemical moieties within LPS may be responsible for inhibition of IP-10 induction by SIN.
Discussion
In the present work, we determined the induction of a select set of chemokines from PBMCs exposed to SIN, as a prelude to understand the virus-induced human innate response. The expression patterns of chemokines and their receptors are important biomarkers for infections and polarization of immune responses (11,36). The levels of CCL2, CCL3, CCL4, and CXCL10 found in SIN-exposed PBMC cultures appear to correlate with those observed in mouse models of virus infection (6). The release of chemokines by SIN-exposed PBMCs in the absence of significant virus replication, mirrors the results obtained from other enveloped viruses (5,33,39). The absence of GFP reporter expression, new viral RNA synthesis as determined by qPCR, and viral particle release, are also suggestive of very poor replication of engineered SIN (Toto1101) in PBMCs. Our results on the chemokine response of PBMCs to SIN particles in the absence of replication are reminiscent of the enveloped virus particle-mediated innate response observed in cultured fibroblasts (7).
The induction of chemokines by viral particles may be type I IFN-dependent or independent, and also appear to be cell-type specific (25). Using a sensitive mRNP-tagging system, Konopka et al. (18) reported that the IP-10 induction in mouse dendritic cells by another alphavirus infection was dependent on IFN-α/β signaling. However, other RNA viruses induce IP-10 synthesis in the absence of type I IFN production and virus replication in fibroblasts (25). A TLR and RIG-1-independent but IRF3-dependent mechanism was proposed for the viral particle-mediated innate immune response (25). Expression of IFN-α and other cytokines from human PBMCs exposed to UV-inactivated viruses indicate immune activation, although expression of IP-10 was not reported (5,33).
In addition to IFN-γ, IFN-α also induces IP-10 synthesis (24). LPS inhibits virus- mediated IFN-α gene expression in human fibroblasts (15). In our experiments, PBMCs exposed to SIN readily produced IFN-α that was also blocked by LPS; however, no IFN-γ was released by SIN (unpublished data). Therefore, it appears that IFN-α signaling could account for IP-10 induction in PBMCs exposed to SIN, since LPS blocked SIN-induced IP-10 release. Notably, Proost et al. (27) reported that IP-10 expression in human PBMCs was induced by dsRNA, and that this induction was blocked by LPS. In our present experiments, IP-10 release occurred in the absence of significant RNA replication, so dsRNA may not be the inducer of IP-10. As indicated previously for fibrobalsts (7,25), the virus particle-mediated immune response appears to account for the chemokine release from PBMCs by SIN. It remains to be established if viral capsid protein can induce IP-10 through activation of NF-κB, as shown for adenovirus (4).
Viral and bacterial co-infections alter the immune response in multiple ways that may be exploited in biomedicine (16). The signaling pathways involved in the host response to viral and bacterial pathogens also appear to overlap (26). The use of LPS and other TLR ligands in SIN infections may help unravel signaling cross-talk between different pathways. Viral RNA can be recognized by TLR3 and 7, and cytoplasmic helicases (16). A TLR7 agonist has recently been reported to downregulate polymicrobial sepsis, suggesting viral infections might have a protective effect against bacterial toxicity (17). The adjuvant activity of IP-10 (19), and its induction by SIN, may strengthen the use of SIN particles in vaccine delivery applications (18). The inhibition of angiogenesis by IP-10 (3) may partly explain the actively pursued oncolytic activity of SIN (37), and thereby promote its potential use in cancer therapeutics.
Footnotes
Acknowledgments
This work was supported by National Institutes of Health (NIH) grant SC1AI081655. We acknowledge the use of MMC Morphology Core and Molecular Biology Core supported in part by NIH grants U54NS041071, G12RR03032, and U54CA91408, RCMI supported Molecular Biology Core facility, AIDS research, and the Vanderbilt-Meharry CTSA grant 1ULRR024975. We thank Nahed Ismail and Fernando Villalta for their critical reading of the manuscript and comments on this work. We thank Ilya Frolov and Guangpu Li for plasmids.
Author Disclosure Statement
No conflicting financial interests exist.