
Abstract
Natural killer (NK) cells have been suggested to play a protective role in HIV disease progression. One potent effector mechanism of NK cells is antibody-dependent cellular cytotoxicity (ADCC) mediated by antiviral antibodies binding to the FcγRIIIa receptor (CD16) on NK cells. We investigated NK cell-mediated ADCC function and the presence of ADCC antibodies in plasma from 20 HIV-1-infected patients and 10 healthy donors. The HIV-positive patients were divided into two groups: six who controlled viremia for at least 8 y without treatment (controllers), and 14 who were persistently viremic and not currently on treatment. Plasma from both patient groups induced NK cell IFN-γ expression and degranulation in response to HIV-1 envelope (Env) gp140-protein-coated cells. Patient antibodies mediating ADCC were largely directed towards the Env V3 loop, as identified by a gp140 protein lacking the V3 loop. Interestingly, in two controllers ADCC-mediating antibodies were more broadly directed to other parts of Env. A high viral load in patients correlated with decreased ADCC-mediated cytolysis of gp140-protein-coated target cells. NK cells from both infected patients and healthy donors degranulated efficiently in the presence of antibody-coated HIV-1-infected Jurkat cells. In conclusion, the character of ADCC-mediating antibodies differed in some controllers compared to viremic patients. NK cell ADCC activity is not compromised in HIV-infected patients.
Introduction
Antibody-dependent cellular cytotoxicity (ADCC) is a humoral immune response mediated by cells of the innate immune system, including granulocytes, macrophages, and NK cells. The Fc portion of antigen-specific antibodies binds to Fc receptors on effector cells and triggers lysis of infected target cells and the secretion of cytokines. NK cells express the activating Fc receptor CD16 (FcγRIIIa), with strongest affinity for the IgG subclasses IgG1 and IgG3 (6,7).
A beneficial function of ADCC has been implicated both in HIV-1-infected patients and in SIV-infected macaques, in which ADCC responses correlate with slower disease progression or lower viral loads (8 –13). Passive transfer of antibodies to macaques established the critical importance of the Fc portion of IgGs in protecting against infection (14 –16). However, ADCC responses in HIV-1-infected patients may be less potent due to deficiencies in antibody affinity for the breadth of viral strains present in the infected individual, and deficiencies of the effector cells. NK cell subpopulations and NK receptor expression in HIV-1-infected patients differ from those in healthy individuals (17 –19), and there are reports of decreased NK cell cytotoxicity and cytokine secretion in HIV-1-infected patients (18,20,21).
The HIV-1 envelope (Env) proteins are expressed on the surface of infected cells and virions, and usually generate a robust antibody response in infected individuals. The functional spike on the virion is a trimeric complex composed of three gp120 and three gp41 proteins, which associate via non-covalent interactions. Monomeric forms of gp120 can be shed from infected cells or virions and induce antibodies often directed to regions that are shielded in the trimeric complex on the virion. When monomeric gp120 and oligomeric Env immunogens have been directly compared in immunization trials the monomeric form has generated less potent neutralizing antibody responses (22,23), indicating that oligomeric forms of the Env protein are preferable when investigating the presence of functional antibodies.
We investigated differences in HIV-1-specific ADCC-inducing antibodies in HIV-1-infected controller and viremic patients. The target proteins used were oligomeric gp140 proteins, which are uncleaved gp120-gp41 complexes in a soluble form (24). In contrast to the proteins or peptides used in previous studies to investigate HIV-1-specific ADCC, this intact Env protein, in a form comparable to that displayed on infected cells, allows detection of ADCC antibodies reacting to both conformation-dependent and relevant linear epitopes. In addition we investigated if NK cells from HIV-1-infected patients and healthy individuals responded differently to antibody-coated HIV-infected cells. We also investigated some factors possibly contributing to the observed heterogeneity in ADCC activity by measuring the ability of V3-loop positive and negative gp140 proteins to elicit ADCC, as well as investigating genotype and NK cell expression level of the ADCC receptor CD16. We found that both controller and viremic HIV-infected patients have ADCC-mediating antibodies in plasma, and that their NK cells have a normal ability to perform ADCC.
Material and Methods
Study subjects
Untreated HIV-1-infected controllers with low viral load, and untreated viremic patients, were recruited (Table 1) (25). As controls, non-HIV-1-infected healthy individuals were included. Ethical permission was received from the regional ethics committee and all participants gave informed consent.
The median (range) for each group is shown.
Three of the viremic patients had previously received antiretroviral treatment (ART), but had not been on ART for the last 3 y prior to sampling.
Two of the controllers had previously been repeatedly vaccinated with gp160 protein. The last vaccination was received more than 7 y ago.
NA, not applicable.
Antibodies and proteins
Anti-human monoclonal antibodies CD56 (NCAM16.2 and B159), CD3 (SK7), CD16 (Leu11a), CD107a (H4A3), and IFN-γ (B27 and 4S.B3) from BD Biosciences (Franklin Lakes, NJ), and CD69 (FN50) from Dako North America (Carpenteria, CA) were used. Isotype control, mouse IgG1, secondary antibody, and anti-mouse IgG/RPE were from Dako. Pooled HIVIG, and purified IgG from Ugandan HIV-1-positive plasma (26) were used to activate NK cells in response to HIV-1-infected cells. HIVIG and the anti-HIV-1 Env antibody 2G12 (AIDS Research and Reference Reagent Program from the National Institute of Allergy and Infectious Diseases, National Institutes of Health: H. Katinger) were positive controls in the RFADCC assay and in gp140 binding ELISAs.
Soluble HIV-1 Env analogues (gp140) from subtype B HIV-1NL4.3 (CXCR4 tropic), HIV-1AD8 (CCR5 tropic), and HIV-1AD8ΔV3 were generated, produced, and purified as previously described (24,27).
Antibody-induced ADCC in the ICS-based ADCC assay
Intracellular cytokine staining (ICS) and degranulation of NK cells activated by HIV-1-specific antibodies in the presence of HIV-1 proteins was previously described (27,28). In brief, whole blood deprived of plasma from a healthy donor was mixed 3:1 with plasma from HIV-1-infected patients or healthy individuals (as a negative control), and gp140 proteins (1 μg/mL) were added. After incubation for 5 h at 37°C, the cells were stained for CD56, CD3, and CD107a, and intracellularly for IFN-γ. Data shown are the net frequency of specifically activated CD56+CD3− NK cells after subtracting the frequency of activated NK cells in the absence of gp140 protein.
Antibody-induced ADCC in the RFADCC assay
In the rapid fluorescent ADCC (RFADCC) killing assay labeled CEM-NKr cells coated with gp140 proteins (5 μg/mL) were incubated for 5 h with plasma from HIV-1-infected patients or healthy individuals, diluted 1:1000, and PBMCs from a healthy donor (27,29). The proportion of lysed target cells (i.e., cells that maintained the membrane dye PKH-26 but lost the cytoplasmic dye CFSE), was determined by flow cytometry. The net frequency of specifically lysed gp140 pulsed target cells was derived by subtracting the frequency of lysed unpulsed target cells.
NK cell-mediated ADCC of HIV-1-infected cells
Jurkat cells were infected with the CXCR4 tropic HIV-1IIIB (LAI) strain at 100× TCID50. At days 7–9 >80% of the cells were positive for HIV-1 p24 (KC57; Beckman Coulter, Brea, CA). Freshly isolated PBMCs from patients or healthy individuals were stimulated with 200 U/mL IL-2 overnight, and then mixed with HIV-1-infected or uninfected Jurkat cells at an E:T ratio of 2:1. HIVIG (250 or 15 μg/mL), anti-human CD107a antibody, and monensin (BD Biosciences) were included in the 4-h incubation. Cells were stained for CD56, CD3, and IFN-γ. Data shown are the net frequency of specifically HIVIG-activated NK cells in the presence of HIV-1-infected Jurkat cells after subtracting the frequency of HIVIG-activated NK cells in the presence of uninfected Jurkat cells.
gp140 ELISA
Half-area plates were coated with 1 μg/mL gp140 protein diluted in sodium carbonate buffer (pH 9.5) overnight. After blocking, plasma from patients and healthy donors were incubated overnight. Goat anti-human IgG (BioRad, Hercules, CA) or mouse anti-human IgG1 (HP6069) or IgG3 (HP6047) from Invitrogen (Carlsbad, CA), all conjugated to HRP, was added. OPD substrate (Sigma-Aldrich, St. Louis, MO) was used to develop the plates at 495–650 nm. End-point titers were determined as the highest titer giving ≥3× the mean OD value obtained for 10 healthy individuals. Half max titers were determined by calculating the dilution at which half of the maximal OD value was derived.
Genotyping
DNA was purified with a QIAamp DNA blood mini-kit (Qiagen, Valencia, CA). A PCR-based method was used to determine CD16 allotypes at position 158 (30). CCR5 genotyping was performed, and one controller, five viremic patients, and one healthy donor were heterozygous for the CCR5Δ32 mutation (25).
Statistical analysis
The following non-parametric tests were used: the Kruskal-Wallis test for differences between multiple groups, the Kruskal-Wallis post-hoc test (Statistica 8.0, Statsoft) to correct for multiple comparisons, the Mann-Whitney U test for differences between two groups, the Wilcoxon matched pairs test for paired analyses, and the Spearman rank test for correlations. Statistical significance was set to p<0.05.
Results
Magnitude and specificity of plasma-induced NK-cell activation
The presence of ADCC-inducing Env antibodies in plasma from viremic and controller patients was investigated with a recently described assay based on ICS (27,28). This assay is less cumbersome than the killing assays, and can measure multiple effector functions of NK cells triggered by HIV-specific antibodies. Gated CD56+CD3− NK cells expressed CD107a and IFN-γ in a gp140-dependent fashion (Fig. 1A), and this was induced by plasma from HIV-positive patients, but not from healthy donors (Fig. 1B and C). Plasma from controllers triggered higher NK cell expression of IFN-γ compared to plasma from viremic patients (Fig. 1C). A similar trend was seen in the frequency of degranulating NK cells, as measured by CD107a staining (Fig. 1B), although this difference was not statistically significant.
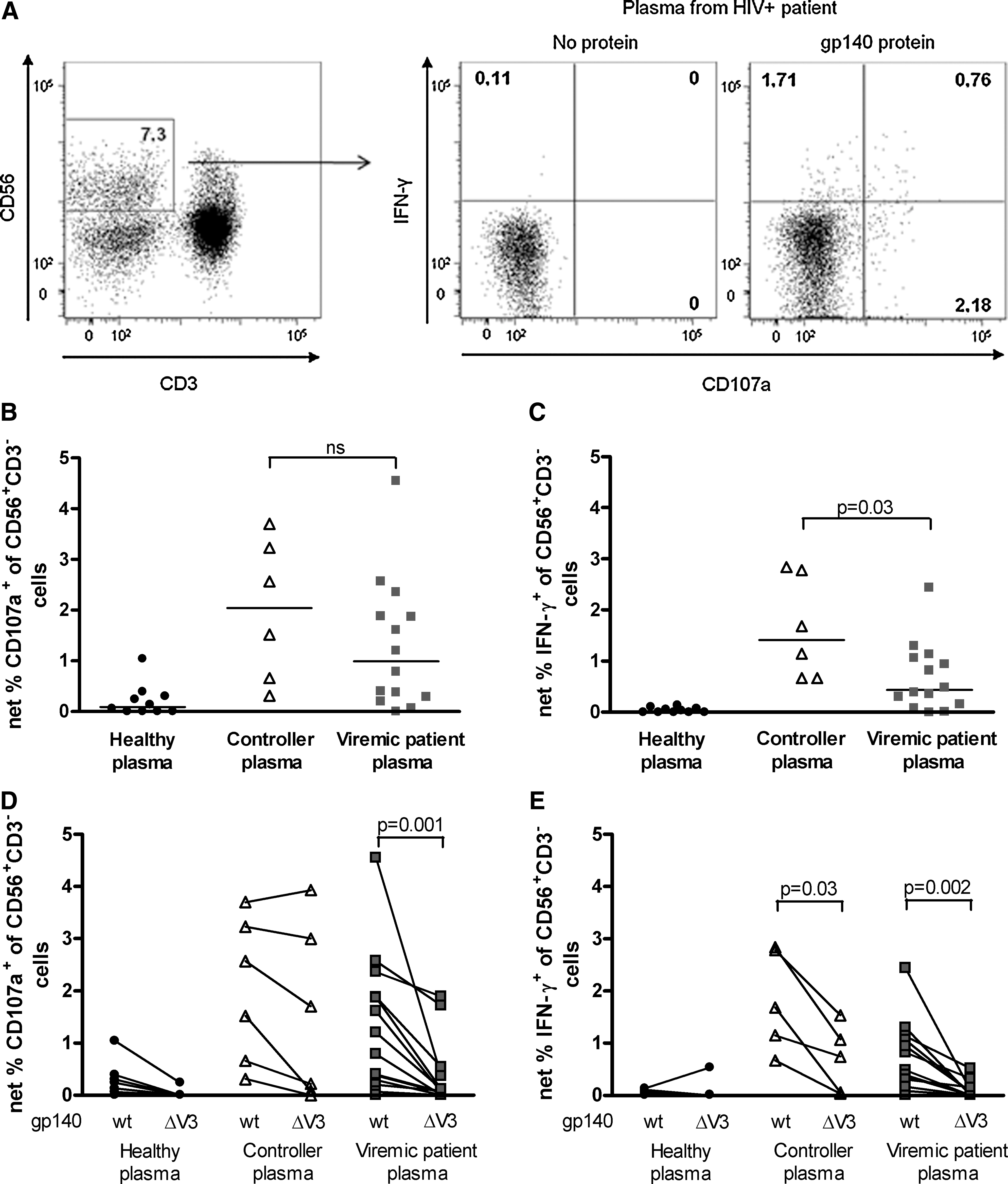
ADCC of patient plasma measured by the ICS-based ADCC assay. (
The V3 loop in the HIV-1 Env protein is a major target for antibodies (31), including ADCC-mediating antibodies (32). It consists of highly variable regions as well as semi-conserved regions important for binding of gp120 to co-receptors (31). To investigate the dependence of the ADCC response to the V3 loop for controllers and viremic patients, a gp140AD8 protein lacking the V3 region was produced (27). Plasma from viremic patients had a significantly reduced ability to induce NK cell degranulation (Fig. 1D), and produce IFN-γ (Fig. 1E) to the truncated protein (ΔV3) compared to the wild-type gp140 (wt) protein. Plasma from controllers also showed a high degree of V3 dependence in NK cell IFN-γ expression (Fig. 1E), whereas degranulation was not significantly affected by deletion of the V3 loop (Fig. 1D). The two controllers with the strongest degranulation to gp140ΔV3 (Fig. 1D) were diagnosed as HIV positive 21 and 24 y ago, respectively, and had a very low viral set point (<50 HIV RNA copies per milliliter). Interestingly, these two controllers were vaccinated with gp160 protein in a vaccination trial which ended 7 y ago (33). Thus, some long-term HIV-1 controllers seem to have V3-independent antibodies inducing NK cell activation, whereas NK-cell-activating antibodies in most HIV-infected patients are largely dependent on the V3 region.
Correlation between the plasma-induced killing of target cells and viral load
In the ICS-based ADCC assay, patient plasma ADCC activity was investigated by determining its ability to activate NK cells in the presence of specific antigen. Historically, ADCC activity of antibodies has been determined by measuring the frequency of target cells killed in the presence of effector cells and antibodies. Therefore, the ability of patient plasma to induce PBMC-mediated killing of labeled CEM-NKr target cells pulsed with HIV-1 gp140 oligomeric proteins was determined by the RFADCC assay (29). Plasma from controllers and viremic patients induced similar killing of gp140-coated targets (data not shown). Of note, there was a statistically significant negative correlation between plasma-induced killing of gp140-coated CEM-NKr cells and viral load in viremic patients, as well as when including all patients (Fig. 2). This indicates that plasma levels of HIV-specific ADCC-mediating antibodies decrease as patients fail to control their viremia.
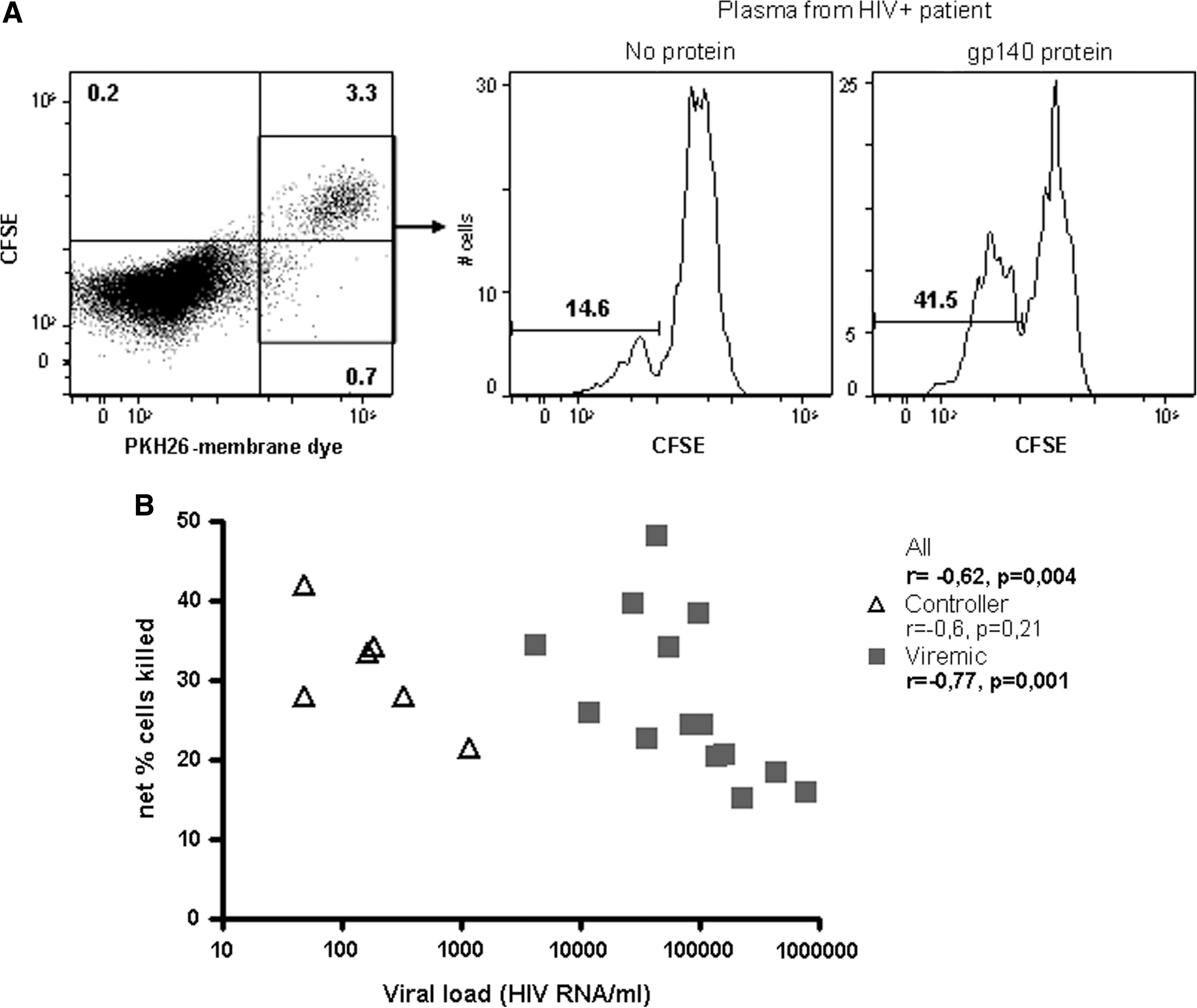
ADCC of patient plasma measured by the RFADCC method. (
Similar IgG, IgG1, and IgG3 concentrations in plasma from controllers and viremic patients
Serum levels of gp140-specific IgG in controllers and viremic patients plasma were determined by ELISA. Oligomeric gp140 proteins from two viral strains, HIV-1AD8 (CCR5 tropic) and HIV-1NL4.3 (CXCR4 tropic), as well as V3 truncated gp140 from HIV-1AD8 (gp140AD8Δ3) were used as target proteins. Levels of gp140-specific total IgG, IgG1 (detectable in all sera), and IgG3 (detectable in 6 of 21 sera) concentrations were similar in viremic patients and controllers, as calculated by end-point titers and half max titers of antibodies to all three proteins (data not shown). There was a statistically significant correlation between IgG1 end-point titers to gp140AD8ΔV3 and the ability of plasma antibodies to induce NK cell degranulation in response to the same protein (r=0.55, p=0.01), while no such correlations were seen for the gp140AD8 wt protein. Thus, gp140-specific IgG1 antibodies that bind to V3-independent regions of the protein may be more important in mediating ADCC.
Decreased levels of NK cells involved in ADCC in viremic patients
For an effective ADCC response in vivo, both specific antibodies of the correct isotype and functional NK cells are needed. Therefore, in addition to determining the ability of patient sera to induce HIV-1 Env-specific ADCC, we characterized the NK cells from the same controller and viremic HIV-1-infected patients. NK cells express the Fc-receptor CD16 (FcγRIIIa), which mediates ADCC (7). Viremic patients had a decreased proportion CD16+CD56+CD3− NK cells in peripheral blood lymphocytes, and a decreased mean fluorescence intensity (MFI) index of CD16 on NK cells compared to healthy individuals (Fig. 3), corroborating previous reports (17,34,35). Controllers showed a similar trend as viremic patients. The finding that both controllers and viremic patients seem to have lower proportions of NK cells that can mediate ADCC indicates that CD16 expression does not contribute to the controller status in our cohort.
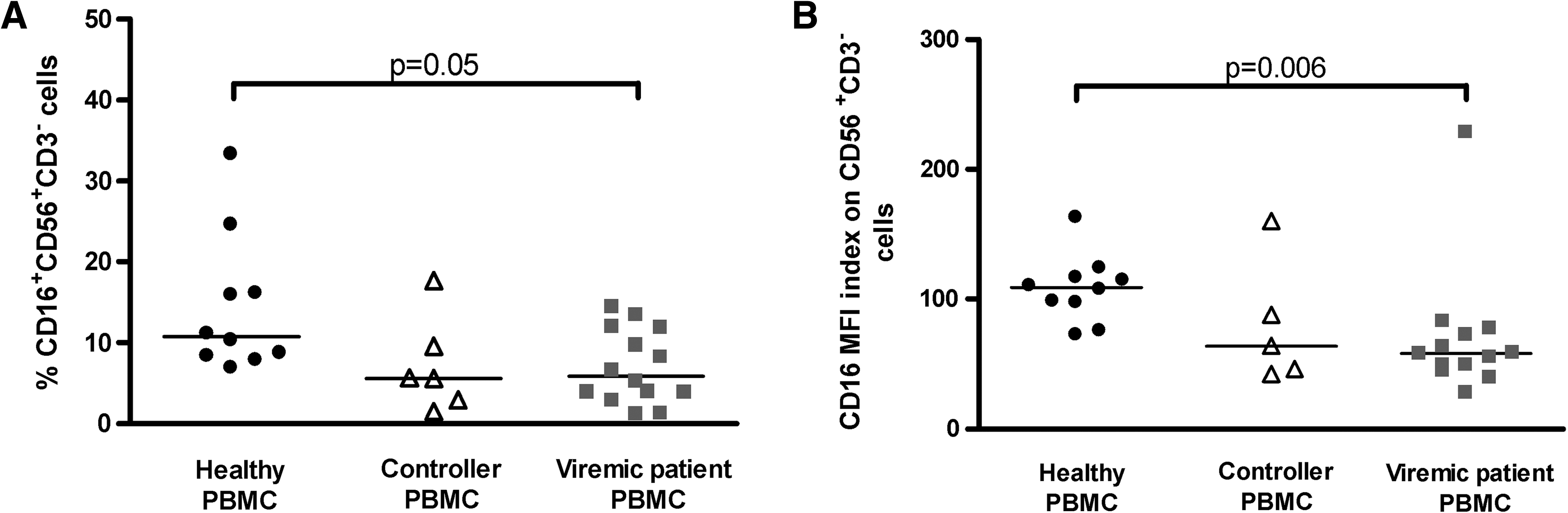
CD16 expression on NK cells. (
Normal NK cell ADCC function in viremic patients
The ability of patient NK cells to degranulate in response to HIV-1-infected Jurkat cells in the presence of HIVIG, pooled IgG from HIV-1-infected patients with high titers of Env-specific antibodies (26), was determined (Fig. 4A). The addition of HIVIG induced activation of NK cells in the presence of both uninfected and HIV-1-infected Jurkat cells. This may be due to the presence of antibodies in HIVIG with specificities to antigens other than HIV. To avoid the influence of this background activity in our analysis of NK-mediated HIV-1-specific ADCC, the fraction of NK cells degranulating in the presence of HIVIG and uninfected cells (median 5%, range 1–12%) was removed from that measured in infected cells. This results in the reported frequency of HIV-1-specific NK cell degranulation or cytokine production in the presence of HIVIG. There were similar frequencies of NK cells degranulating and expressing IFN-γ in HIV-1-infected patients and healthy individuals (Fig. 4B and C). These results indicate that although the NK cell expression of CD16 was lower in viremic patients compared to healthy donors (Fig. 3B), their NK cells are not defective in their capacity to become activated in response to HIV-1-specific antibodies. However, HIV-1-infected patients have a lower frequency of NK cells in peripheral blood that can exert this ADCC function (Fig. 3A).
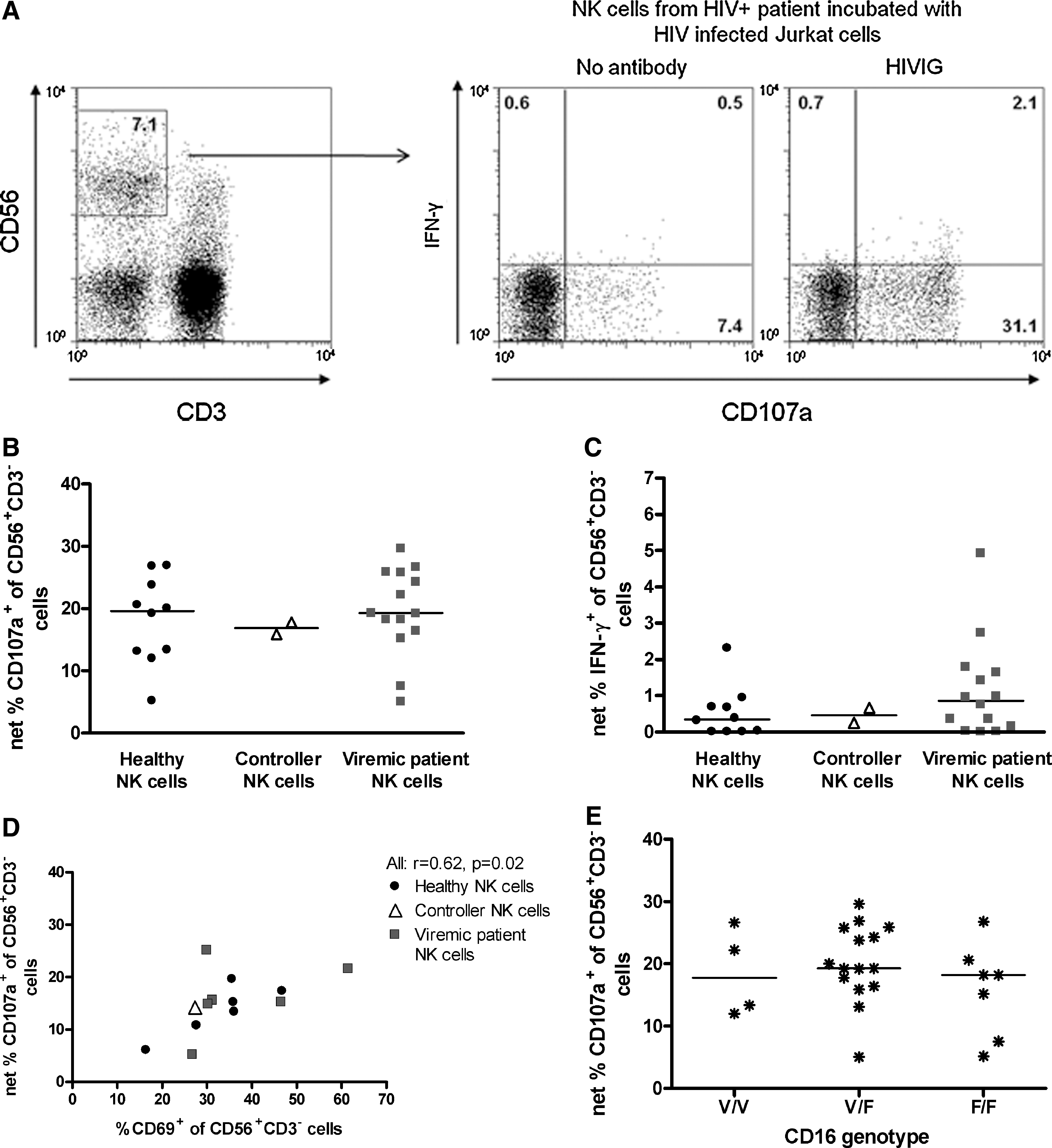
ADCC mediated by patient NK cells. (
Ex vivo expression of CD69 and CD16 correlates with HIV-1-specific NK cell ADCC function
Despite the lack of difference between patients and healthy donors in HIV-1-specific NK cell ADCC activity, there was quite a range in the degree of responses (Fig. 4B and C). We hypothesized that the ability of NK cells to respond to HIVIG may be explained by differences in NK cell phenotype, and investigated whether ex vivo expression of the Fc-receptor CD16 correlated with the HIV-1-specific NK cell ADCC activity. As expected, the frequency of CD56+CD3− NK cells expressing CD16 correlated with the frequency of NK cells degranulating in the presence of HIVIG (r=0.6, p=0.02). Similarly, the frequency of NK cells expressing the activation marker CD69 ex vivo correlated significantly with the proportion of NK cells degranulating in the presence of HIVIG (Fig. 4D). Thus, in vivo activated CD16-expressing NK cells may be more efficient in mediating ADCC.
CD16 genotype does not influence HIV-1-specific NK cell ADCC function
There are amino acid variations in Fc receptors that cause differences in ADCC efficacy. One such variation is found in CD16 (FcγRIIIa) at position 158, where either a valine (V, high ADCC) or a phenylalanine (F, low ADCC) is present (6,36). Genotyping at this position revealed similar frequencies of the various genotypes in healthy individuals, controllers, and viremic patients (data not shown). There were no differences in HIV-1-specific NK cell activation in the presence of HIVIG between individuals homozygous for V or F, or heterozygous with both V and F (Fig. 4E). Thus, reported differences between genotypes did not have a major influence on NK-cell ADCC activity in this assay and cohort.
Discussion
We characterized ADCC-mediating antibodies in plasma from HIV-1-infected patients, controllers, and viremic patients. We found that plasma from both groups of patients contained HIV-specific antibodies mediating ADCC. In viremic patients and in some of the controllers, much of the HIV-1 gp140-specific ADCC effect was dependent on the V3 region of the protein (Fig. 1D and E), corroborating previous reports (27,32,37). Two of the patients who controlled HIV infection had a similar response to the wt gp140 and the gp140ΔV3 proteins (Fig. 1D), and are likely to have ADCC-inducing antibodies with a broader specificity directed to other parts of the gp140 protein. Interestingly, these two controllers were included in a gp160 vaccination trial that ended more than 7 y ago (33). Despite the long time since the last vaccination, it is possible that the vaccinations contributed to the development of antibodies directed to other parts of the Env protein. One possible explanation to the lower reactivity to gp140ΔV3 compared to wt gp140 is that deleting the V3 region could have distal conformational effects on the protein, affecting the integrity and/or exposure of non-V3 epitopes, and thus the binding of plasma antibodies. On the other hand, it is possible that we have underestimated the V3-specific ADCC response, as we have used gp140 of only one strain, AD8. ADCC activity to each patient's homologous strains may be higher. To our knowledge this is the first report comparing differences in specificity of plasma ADCC-mediating antibodies between viremic patients and controllers. Such differences should be further investigated in larger patient cohorts.
Although we detected a slightly elevated NK cell production of IFN-γ in controllers compared to viremic patients, we could not detect elevated NK cell degranulation or increased cytolysis induced by plasma from controllers compared to viremic patients. In contrast, a previous study of elite controllers reported an increased ability to induce killing of gp120-coated target cells (11). One difference between the studies is the antigen used. While we used gp140, the study by Lambotte et al. used monomeric gp120, which lacks the epitopes of gp41, and probably differs in exposure of gp120 epitopes due to a lack of a quaternary structure compared to gp140. In addition, epitopes of the V3 loop may be better exposed in gp140 compared to gp120 (38). Different controller patients may have various reasons for their controlling activity, for example host genetic factors, variation of viral replication capacity, or strong ADCC antibody responses. Thus, competent ADCC may be important for some controllers but not for others.
Our correlation between increased plasma-mediated ADCC and lower viral load in patients (Fig. 2) corroborate previous reports in a variety of settings (9,39,40). Our data also adds to the growing interest in the role of ADCC in controlling or preventing HIV infection. It has been speculated that ADCC played a role in the modest but statistically significant protective effect seen in the Thai RV144 vaccination trial (41), since the vaccine regimen elicits robust ADCC activity (42).
In addition to studying HIV-specific ADCC-mediating antibodies in patient plasma, we also analyzed ADCC function of the patients' NK cells. There are contradictory reports concerning the ability of effector cells from HIV-1-infected patients to mediate ADCC; some report normal ADCC activity (43,44), whereas others suggest a correlation between decreased effector cell-mediated ADCC activity and disease progression (35,45,46). Most previous studies have used the frequency of target cells killed in the presence of patient PBMCs as a read-out (43 –46). It is known that viremic HIV-1-infected patients have a lower frequency of CD56+CD16+ NK cells (corroborated here, Fig. 3A), and an increased fraction of dysfunctional CD56−CD16+ NK cells compared to healthy individuals (17,19). Both are factors that are likely to influence the frequency of target cells killed by unfractionated PBMCs. This is the first study comparing the activity of CD56+CD3− NK cells from viremic patients and healthy individuals in an ADCC assay based on HIV-infected target cells. We found that NK cells from viremic patients and healthy individuals were similarly activated to degranulate and secrete IFN-γ in HIV-1-specific ADCC (Fig. 4B and C), as well as in response to K562 target cells (25). This suggests relatively normal NK cell function at a cellular level in HIV-infected subjects. In two recent studies the frequency of ADCC-induced NK cell degranulation in response to antibody-coated mouse leukemic cells or CD16 cross-linking was lower in viremic patients compared to healthy individuals (34,35). It is possible that the use of different experimental systems to induce ADCC contribute to the different conclusions reached.
Despite the similarities in ADCC activity seen between HIV-1-infected patients and healthy individuals, viremic patients had a decreased level of expression of the Fc-receptor CD16 on NK cells (Fig. 3B), corroborating previous reports (17,35). Possible factors in HIV-1-infected patients that might contribute to the decreased expression of CD16 on NK cells include defects in expression of the ζ chain associated with CD16 (47), and the recently reported increased expression of matrix metalloproteinases that removes CD16 from the cell surface following NK cell activation (35). The positive correlation between ADCC-induced NK cell degranulation and the frequency of NK cells expressing CD69 ex vivo (Fig. 4D) might be due to the proposed function of CD69 as a stimulatory receptor inducing NK cell-mediated cytotoxicity when triggered (48), or could be a marker of in vivo activation of NK cells.
Many factors might influence ADCC responses to HIV-1-infected cells in vivo, including antibody specificity and cellular functions, and to characterize these parts it may be necessary to use different assays to measure ADCC activity. Further investigations are warranted to delineate the importance of ADCC immune responses in HIV-1-infected patients, and to identify epitope specificity of ADCC-inducing antibodies in patients that can control their disease. Such findings should assist future therapeutic and preventive developments based on ADCC.
Footnotes
Acknowledgments
We thank Professor Jan G.J. van de Winkel, Jeanette Leusen, and Miranda Blokland, from the Department of Immunology at the University Medical Center Utrecht, The Netherlands, for protocol and control samples for the CD16 genotyping.
This research was supported by the Swedish Research Council, Swedish International Development Cooperation Agency (SIDA), and The Swedish Physicians Against AIDS Research Foundation.
Author Disclosure Statement
No competing financial interests exist.