
Abstract
Within the past decade, human infections with the highly pathogenic avian influenza H5N1 have resulted in approximately 60% mortality and increased the need for vaccines and therapeutics. Understanding the molecular events associated with pathology can aid this effort; therefore, this study was conducted to assess microRNA (miRNA) expression in mouse lungs infected with H5N1 A/Vietnam/1203/04. Intranasal administration of 1500 median tissue culture infectious dose of H5N1 promoted differences in the number and expression pattern of miRNA from lung tissue collected at 2, 4, 6, 24, and 96 h post-exposure that mapped to common biological functions. Informatics analysis identified miRNA-specific predicted genes known to be therapeutic drug targets in which Furin was common to all time periods. This study provides insight into the differential miRNA expression with respect to the host-pathogen relationship and identification of potential therapeutic drug targets.
Introduction
Understanding the underlying molecular mechanisms that control tissue responses to external insults can provide important steps towards the development and testing of treatments for existing and emerging infectious diseases. The careful control of biological systems by cellular protein effectors and subsequent changes in normal functions would be expected to be reflected in the process of increased or decreased mRNA expression and protein synthesis. Commercial gene expression technologies enable the characterization of thousands of transcripts from a single tissue sample; therefore, the level of mRNA transcripts in normal and diseased tissue should provide some indication of the relative proportion and importance of each of these functions. Monitoring and analyzing gene expression profiles can provide valuable insight into the development of prophylactic and/or therapeutic treatments targeting specific molecular pathways or genes that can reduce the risk and severity of disease.
MicroRNAs (miRNAs) are endogenous short (∼22 nucleotides) non-coding RNAs that regulate gene expression post-transcriptionally by modulating the stability and translation of mRNAs. Post-transcriptionally, each miRNA has the potential to regulate more than 100 different mRNA transcripts, resulting in a 20–30% alteration of protein-encoding genes (9). MicroRNAs have recently received attention with respect to their potential role as indicators of disease and diagnosis for various cancers as well as host-pathogen interactions (9 –16). An important goal for analyzing changes in miRNA expression patterns could be to design novel diagnostic and therapeutic strategies against pathogens based on their roles as effectors of biological and cellular processes. Therefore, the goal of this study was to perform a preliminary assessment of miRNA expression in lung tissue of mice infected with H5N1 A/Vietnam/1203/04 to provide information regarding molecular events associated with pathogenesis. The time periods selected for this study were intended to identify changes in miRNA expression that occur during initial infection and disease progression. These include: (1) the initial stages of virus binding, uptake, and replication prior to release of progeny virus (2, 4, and 6 h post-infection), (2) following the potential for multiple rounds of viral replication, increasing tissue viral load, and initial host response (24 h post-infection), and (3) the late- or end-stage of the disease process (96 h post-infection).
Materials and Methods
Influenza exposure
All work was conducted under biosafety level 3 (BSL-3) conditions. Female BALB/C mice (5–6 wk of age) were purchased from Charles River Laboratories (Wilmington, MA). The mice were maintained under an animal care and use program accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. The animals were group-housed and provided potable water and feed ad libitum. The animals were maintained on a 12-h light/dark cycle with no twilight. Air temperature in the animal rooms was maintained within the range 16 to 27°C, with relative humidity maintained between 30 and 70%.
The highly pathogenic avian influenza A/Vietnam/1203/04 virus was propagated in the allantoic cavity of 10-day-old hen eggs at 37°C. The allantoic fluid from infected eggs was harvested and stored at −70°C. The A/Vietnam/1203/04 virus used for challenges was passed a single time in embryonated hen eggs, and the concentration of virus was determined to be 1.4×108 tissue culture infectious dose of 50% (TCID50)/mL. This viral stock was diluted in phosphate-buffered saline (PBS) to 3×104 TCID50/mL. The animals were challenged via the intranasal route in a total volume of 50 μL, corresponding to 1500 TCID50 of virus. Control groups received allantoic fluid diluted in PBS similar to the virus stock and administered intranasally in a 50-μL volume. All mice were placed in filtered caging by group following challenge.
Sample collection, RNA isolation, cDNA synthesis, and hybridization
At 2, 4, 6, 24, and 96 h post-challenge, animals were anesthetized and terminally bled by cardiac stick. The lungs were removed and stored in RNAlater ® until processing for miRNA profiling. Total RNA was isolated from individual infected lung samples (n=3/time-point) and a control group (n=3). Each lung sample was homogenized in QIAzol Lysis Reagent (Qiagen, Valencia, CA) using Soft Tissue Omni Homogenizer Tips (Omni International, Marietta, GA). Following homogenization, chloroform was added to each sample, the aqueous and organic phases were separated by centrifugation, and miRNA was isolated using the Qiagen miRNeasy Mini Kit. Total miRNA for each sample was evaluated for concentration and purity using a NanoDrop ND-1000 Spectrophotometer (NanoDrop, Wilmington, DE), and integrity by gel electrophoresis using the FlashGel® RNA cassette system (Lonza, Rockland, ME).
miRNA and transcript analysis
Total miRNA (100 ng) was labeled using the FlashTag™ Biotin RNA Labeling Kit (Genisphere, Hatfield, PA). In brief, the labeling process consisted of a tailing reaction followed by ligation of the biotinylated signal molecule to the target miRNA samples. The labeled miRNA was then hybridized to an Affymetrix GeneChip® miRNA array (46,228 probe sets, 6703 miRNA sequences, 71 organisms, 609 mouse miRNA; Affymetrix, Santa Clara, CA). Hybridization, washing, and staining were performed according to the Affymetrix recommended protocols. After scanning, the digitized image data was processed using GeneChip® Command Console® Software (Affymetrix). Quality control analysis was performed using the miRNA QC Tool (Affymetrix) and analyzed using the Partek Genomics Suite (Partek, St Louis, MO).
After analysis of the report file for each chip and confirmation that the chip image was not smeared or distorted, each Affymetrix “.CEL” file was imported into the Partek Genomics Suite. An analysis of variance (ANOVA) with Tukey's HSD post-hoc analysis, including a multiple testing correction (Benjamini and Hochberg false discovery rate set at p≤0.05) was used to generate a list of all significantly-changed (p≤0.05) miRNAs for each time-point. In order to not exclude any significantly-changed miRNA that may exhibit potential biological importance, no fold-change cut-off was assigned for these samples. Thus, all significantly-changed miRNA data were used for the informatics analysis to identify predicted gene targets.
Biological function analysis and predicted gene targets
Lists of miRNA from each time-point were transferred from the Partek Genomics Suite into Ingenuity Pathways Analysis (IPA) 8.5 (Ingenuity Systems,

Analysis strategy of differentially expressed miRNAs using IPA. Lists of significantly-changed (p≤0.05) miRNAs at each time-point were imported into IPA and mapped. Biological function analysis was performed separately on all time-points (2, 4, 6, 24, and 96 h post-exposure) containing significantly-changed genes (
At each time-point, miRNA lists within IPA were used to generate a list of predicted gene targets. To do this, each miRNA list was imported into a single, custom “grow pathway” function to identify genes that possess either direct or indirect relationships. Once each list of miRNA gene targets was identified, IPA was used to identify drugs within the IPA knowledge base that were in Phase II or Phase III clinical trials or FDA-approved for use against specific genes. To generate a list of potential drugs, a single list containing genes at each time-point was imported into a single, custom “build pathway” function within IPA. Once all genes were imported into a single pathway data set, the “drug overlay” function was implemented that yielded all drugs within the IPA knowledge base.
Results
miRNA analysis
Statistical analysis of expression data revealed temporal differences in the number and expression pattern (i.e., increased or decreased) of miRNA from lung tissue exposed to A/Vietnam/1203/04 (Tables 1 –5). The greatest number of significantly changed (p≤0.05) miRNAs was observed at 6 h post-infection when 38 miRNAs were increased and 21 miRNAs were decreased (Table 3). Fig. 2 shows the number of significantly (p≤0.05) upregulated and downregulated miRNAs over the 2-, 4-, 6-, 24-, and 96-h time periods.

Number of significantly upregulated and downregulated miRNAs at 2, 4, 6, 24, and 96 h post-exposure to H5N1 A/Vietnam/1203/04 compared to controls.
Biological function analysis and predicted gene targets
The biological functions analyzed using IPA were categorized in order based on the level of statistical significance for each experimental group. Over the observed time periods evaluated in this study, the type and number of significantly changed biological functions varied from 4 to 20 (Tables 6 –10). Among these experimental time points, three common biological functions were commonly shared, and included Cancer, Reproductive System Disease, and Cellular Growth and Proliferation.
To provide a better understanding of the roles of miRNAs expressed over time in H5N1-infected lung tissue, IPA was used to generate gene lists known to have direct or indirect functional relationships with these miRNAs. Using this approach, the number of predicted genes varied over time, with 323 genes identified at 2 h, and a maximum of 724 genes observed at 6 h (Fig. 3). Moreover, predicted genes within these lists were sorted based on those having drugs within the IPA knowledge base that were in Phase II or Phase III clinical trials or FDA-approved for use against specific gene products (Tables 6 –10). The number of these predicted genes varied over the observed time period, and 7 were observed at 2 h (Table 6), and a maximum of 32 were identified at 4 h (Table 7). The gene encoding furin (a subtilisin-like proprotein convertase) was the only gene predicted to be regulated by the miRNAs identified in the study over the 2-, 4-, 6-, 24-, and 96-h time periods. However, there were other predicted genes (BCL2, CASP3, and IL-6) that were common in at least 3 of the 5 time-points.
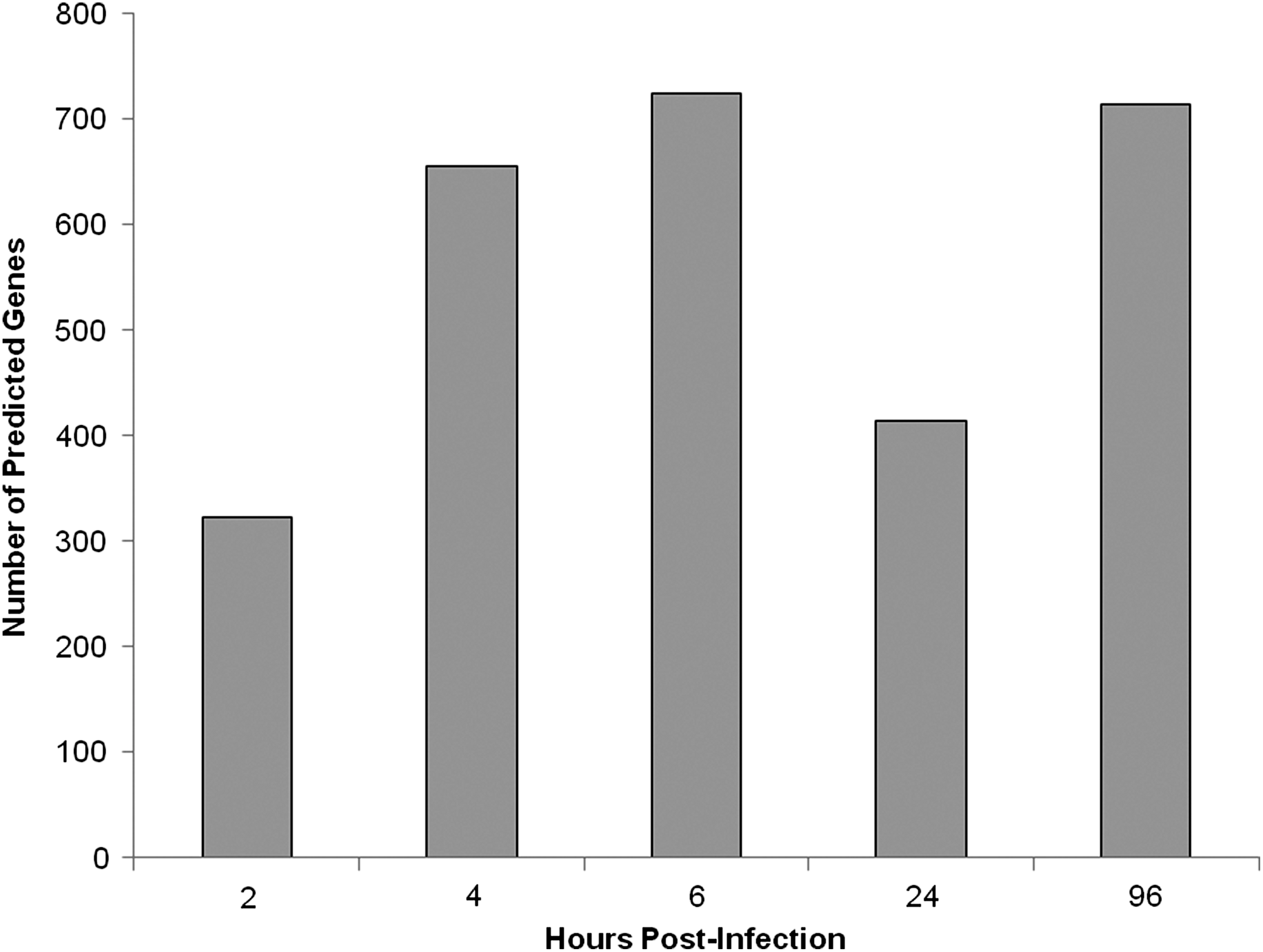
Number of predicted genes having direct or indirect relationships with miRNAs identified using the IPA knowledge base at 2, 4, 6, 24, and 96 h post-exposure.
Discussion
Studies evaluating host-pathogen responses are important to understanding the mechanistic relationship of infection, resistance, and disease progression. Such studies include the use of both in vitro and in vivo models that encompass clinical, pathological, cellular, and molecular end-points. The present study provides an initial assessment of differential miRNA expression in mouse lung tissue infected with H5N1 A/Vietnam/1203/04. As miRNAs are becoming more prevalent as molecular and physiological effectors of disease, the data from this study could ultimately be used to aid in the understanding of the underlying molecular mechanisms associated with H5N1 A/Vietnam/1203/04 pathogenicity for developing or evaluating vaccines and therapeutics.
The characterization of miRNA expression patterns associated with influenza infection have provided insight into the cellular pathways associated with virulence and host-pathogen interactions, including immune responses and cell death (11,16). In the present study, mice infected with H5N1 A/Vietnam/1203/04 exhibited significant changes in the biological functions, providing a higher-level glimpse at the host-pathogen relationship with respect to differential miRNA expression. These observed changes in biological functions are underscored by similar patterns of significant miRNA expression, as well as the predicted gene targets that are known to possess either direct or indirect relationships. Since a single miRNA has the potential to regulate the expression (induce or suppress) of hundreds of different mRNA transcripts, sorting through known miRNA-mRNA relationships is one approach to help identify potential therapeutic gene targets. Among the genes that encode proteins known to be modulated by drugs/compounds, furin was the only gene identified at each of the time-points sampled in the current study. Furin is a member of the proprotein convertase family that activates precursor proteins by cleavage of multibasic residue sequences, and plays an important role in many physiological and pathological processes (17). Furin has been shown to participate in the cleavage and activation of bacterial toxins and viral proteins that leads to the processes resulting in viral pathogenesis and toxin activity (17 –21). Moreover, furin-mediated cleavage of hemagglutinin (HA) is an important step in the sequence of events leading to H5N1 pathogenicity (18,19,21).
Using the IPA knowledge base to identify drugs that are in Phase II or Phase III clinical trials or FDA-approved, the compounds hexa-D-arginine amide (D6R) and nona-D-arginine amide (D9R) were found to target furin. Polyarginine compounds, such as D6R and D9R, are inhibitors of furin that exhibit little toxicity (22). Both D6R and D9R have been used as therapeutic treatments to protect against the diseases and pathologies caused by bacterial toxins and viruses (23 –26). Based on the identification of furin as a predicted gene regulated by the miRNAs observed in this study and previous work of other investigators, it appears that furin would be a potential therapeutic target upon which to focus future research efforts. Such a strategy for targeting cellular processing proteases of HA has been suggested as a promising anti-influenza therapeutic approach (19,26).
It should be noted that the observed changes in miRNA expression described in this study were comprised of an miRNA pool isolated from multiple cell types that comprise each individual piece of mouse lung tissue. Therefore, the observed results can be assumed to represent an average of the miRNA expression changes in these multiple cell types within the lungs. Further investigation into the regulation and effects of specific molecular pathways and genes needs to be done to identify diagnostic markers and potential therapeutic targets for treating H5N1 pulmonary infections. The molecular networks and genes that are unique or specific to H5N1 infection should be the focus, as these differences may prove to be more beneficial. Moreover, further verification of changes in expression for key miRNAs should be performed to strengthen the argument of using miRNAs and subsequently-affected proteins as therapeutic targets. Ultimately, the correlation of miRNA, mRNA, and protein expression from the same H5N1-infected lung samples may help in characterizing an overall pattern of signaling events and changes in the host response that would be beneficial to identifying molecular targets for new therapies against lethal H5N1 infection.
Footnotes
Acknowledgment
This work was funded by Battelle's Independent Research and Development Program.
Author Disclosure Statement
No competing financial interests exist.