
Abstract
Clinical trials with highly-active antiretroviral therapy (HAART) have shown that a substantial number of patients continue to show a decrease in viral load and/or increase or stable CD4+ T-cell numbers even in the presence of multidrug resistant (MDR) viruses. We compared replication capacity (RC) and expression of anti-apoptosis marker genes (AAMGs) in human peripheral blood mononuclear (PBM) cells infected with NL4-3 (wild-type; WT) and mutant viruses. Replication kinetics assays showed a significant decrease in RC of all mutant viruses in comparison to the WT virus. The viruses containing patient-derived MDR RT without the K65R mutation (PSD5.2) replicated efficiently in comparison to the viruses with MDR RT containing the K65R mutation (PSD5.1), or the single mutations K65R and M184V. Compared with WT, a significant decrease in RCs of viruses: K65R (RC=0.39±0.02; p≤0.0001), M184V (RC=0.72±0.04; p≤0.0001), PSD5.1 (RC=0.32±0.04; p≤0.0001), and PSD5.2 (RC=0.90±0.04; p=0.002) was observed on day 10. RT-PCR-based apoptosis array was performed on total cellular RNA. Recombinant virus PSD5.2 showed a 1.5- to 6-fold upregulation in 8 AAMGs (AKT1, BAG3, BCL2A1, BFAR, BIRC2, BNIP1, BNIP3, and CFLAR) on day 1 and day 7 post-infection with respect to WT virus. PSD5.1 showed upregulation of only one gene (BAG1) on day 1 (1.75-fold) and day 7 (1.97-fold). Point mutant K65R showed a 1.5- to 4-fold upregulation of six AAMGs on day 7. Viruses with the M184V mutation showed upregulation of only one gene (BAG1). These observations indicate that the upregulation of specific AAMGs may not be dependent on the RCs of HIV-I variants, and that the possible interaction among mutated RT residues and viral and/or host proteins may induce CD4+ T-cell-protective anti-apoptosis proteins.
Introduction
Both in vivo and in vitro studies have demonstrated that CD4+ T cells and the predominantly peripheral CD4+ T cells deplete over the course of HIV-1 infection as a consequence of both HIV-1 and by-stander mechanisms of apoptosis induction (5,29,32). On the other hand, the predominantly uninfected peripheral monocyte pool (44) does not diminish during steady-state viral replication (38,45). A recent study by Giri et al. (26), using monocytes from peripheral blood mononuclear (PBM) cells of HIV-infected subjects, demonstrated the presence of a stable anti-apoptosis gene signature comprised of 38 genes associated with p53, CD40L, TNF, and MAPK signaling networks. They further showed that this gene signature is associated with cadmium chloride- or Fas ligand-induced apoptosis resistance in circulating monocytes, in contrast to increasing apoptosis in CD4 T cells from the same infected subjects (26).
CD4+ T-cell apoptosis is a dynamic process and involves a series of cytokines/chemokines (13,22,35) and pro- and anti-apoptosis-inducing proteins (3,9,31,41,59). Based on the aforementioned fact, we speculated that HAART-selected MDR viruses may be involved in the upregulation of specific anti-apoptosis marker genes (AAMGs), and thereof anti-apoptosis marker proteins (AAMPs). We compared replication capacity (RC) of HIV RT variants and performed an RT-PCR-based apoptosis array on total cellular RNA isolated from PBM cells infected with WT (NL4-3) and HIV RT variants. We demonstrate here that specific AAMGs were upregulated in mutated viruses in comparison to WT virus, and that the expression of these genes was independent of the RC of the viruses. To our knowledge this is the first study to compare expression of AAMGs in relation to RCs of HIV RT variants.
Materials and Methods
Chemicals and medium
The radionucleotides (methyl-3H)dTTP was purchased from Perkin Elmer (Shelton, CT), and polynucleotide poly(rA) and primer oligo(dT)12–18 were purchased from Invitrogen (Carlsbad, CA). The oligonucleotides used for mutagenesis were synthesized and high-pressure liquid chromatography was purified by Diversified Biopharma Solutions Inc. (Loma Linda, CA). Complete Dulbecco's modified Eagle's medium (DMEM) containing 10% heat-inactivated fetal bovine serum (FBS) and penicillin/streptomycin was used to grow 293T cells. Complete RPMI medium containing 20% FBS, 26 IU/mL of IL-2, penicillin/streptomycin, and glutamine was used to culture PBM cells.
Cells and virus
Blood from an HIV-infected veteran (PSD5) was collected in a BD Vacutainer CPT (Becton Dickinson, Franklin Lakes, NJ) after IRB approval and proper informed consent. Healthy donor PBM cells were prepared from buffy coats received from commercial vendors (Red Cross and LifeSouth Community Blood Center, Atlanta, GA) using Ficoll gradients. Primary human embryonic kidney cells 293T, indicator cell line HeLa-CD4-LTR-β-galactosidase (34), and proviral clone pNL4-3 (1), were obtained from the AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Site-specific mutagenesis
Single-point mutants K65R and M184V were created in the background of proviral clone pNL4-3 by using the pALTER−1 mutagenesis system of Promega (Madison, WI) according to the manufacturer's guidelines and our previously described protocols (51–54; Table 1).
Site-directed point mutants K65R and M184V were created in NL4-3 vector (1) by exchanging a 4.3-kb Sph1-Sal1 wild-type fragment with mutated fragment from pALTER mutagenesis vector. RT fragments (1–250 codons) of MDR clinical isolate (PSD5) were cloned into pNΔRTPR vector (6). resulting in ΔRT5.1 (PSD5.1) and ΔRT5.2 (PSD5.2) proviral clones.
Preparation of recombinant viruses containing MDR RT of clinical isolate
RNA was isolated from the patient's (PSD5) plasma and RT-PCR was performed using previously described primers: RTA1 5′AATTTTCCCATTAGTCCTATT3′ and Gag3 5′TAAGTCTTTTGATGGGTCATAATA3′ (8). Population cloning was performed in Topo TA cloning vector PCRR2.1 (Carlsbad, CA), and sequencing was performed to analyze linkage among mutations. Out of 11 clones tested, 1 had mutations K65R, D67N, S68G, K70R, K103N, T215Y, and K219E, and 10 clones had all these mutations except K65R. In order to sub-clone RT fragments in RT deletion vector pNΔRTPR, amplification of RT fragments (1–250 amino acids) containing K65R (PSD5.1) and without K65R (PSD5.2) was performed using previously described primers: pH10 5′TTTCCCATTAGTCCT
Virus production
Viruses were produced using SuperFect reagent (Qiagen, Valencia, CA) per the manufacturer's guidelines. Cells (293T) were split into 60×10-mm dishes 24–48 h prior to transfection. To generate virus the complex containing 10 μg of DNA in 150 μL of serum-free medium and 30 μL of SuperFect reagent was incubated at room temperature for 10 min. One milliliter of complete DMEM was added drop by drop onto 293 cells that were washed once with phosphate-buffered saline (PBS). Cells were incubated at 37°C in the presence of 5% CO2 for 3 h. The remaining medium-complex was removed and the cells were washed with 4 mL of PBS. Four milliliters of complete DMEM was added and dishes were incubated for 72–96 h. Culture supernatants were collected and centrifuged for 5 min at 833 g (g=1.2) to pellet any debris. Culture supernatants were filtered (0.22 μm) and saved in aliquots of 0.5 mL and 1 mL at −80°C.
Quantification of virus
Both HIV-1 antigen p24 concentrations as well as RT activity for each stock virus were determined as described previously (11,12,51). Briefly, antigen p24 determination was done according to the manufacturer's protocol using the Antigen p24CA ELISA kit (NCI, Frederick, MD). To determine RT activity, 1 mL of each virus was centrifuged for 2 h at 15,000 rpm in a refrigerated centrifuge (Biofuge 15R, Rotor 3743; Heraeus Instruments Corp., South Plainfield NJ). Pelleted virions were lysed with 50–100 μL of virus solubilization buffer (0.5% Triton X-100, 50 mM Tris, pH 7.8, 800 mM NaCl, 0.5 mM PMSF, and 20% glycerol), 10 μL of samples in triplicate were mixed with 75 μL of RT assay buffer (60 mM Tris, pH 7.8, 12 mM MgCl2, 6 mM dithiothreitol, and 7 μg dATP) in the presence of 450 ng of poly (rA)-Oligo (dT) and 5 μCi of methyl-3H TTP, and the reactions were incubated at 37°C for 2 h. The entire reaction mixture was overlaid on a DE81 filter. The filters were washed 3 times with 2× SSC buffer, 2 times with absolute alcohol, air dried, and the radioactivity was measured in scintillation fluid.
Determination of viral titer
Viruses produced in 293T cells were quantified for TCID50 using a ACTG/DoD protocol described elsewhere (33), and multiplicity of infection (MOI) was calculated using infecting HeLa-CD4-LTR-β-galactosidase cell lines as described elsewhere (34).
Replication kinetics assays
Replication kinetics assays were performed as described elsewhere (51,52). Briefly, 10×106 PHA-stimulated PBM cells were infected with an equivalent amount (100 TCID50/106) of viruses. Culture supernatants were collected every other day until day 10 to determine antigen p24 concentration, RT activity, and genomic RNA sequence. Replication capacity was calculated by dividing percentage of antigen p24 production by mutant virus in relation to WT virus (RC=M/W, where M is the percentage of antigen p24 production by mutant virus, and W is the percentage of antigen p24 production by WT virus).
Preparation of RNA
Viral RNA was isolated by the QiAamp® viral RNA mini kit (Qiagen). RNA integrity (RIN) was assessed with an Agilent Bioanalyzer. A RIN of >5 was considered for PCR reaction. RT-PCR was performed using the Superscript™ III one-step RT-PCR system (Invitrogen). All the stock viruses were confirmed by sequencing viral RNA using primer 74F, 5′-GTAGGACCTACACCTG TCAAC-3′ (12).
Apoptosis array analysis
PHA-stimulated PBM cells were infected with 0.001 MOI of each virus and infected cells were collected on day 1 and day 7 post-infection. Cells were washed twice with PBS and stored at −80°C until use. Total cellular RNA was isolated as discussed above. A reverse transcription reaction, using 150 ng of total RNA, was done with the Qiagen RT2 First Strand Kit (cat. no. 330401). cDNA samples were then assayed using the Apoptosis RT2 Profiler PCR Array (Qiagen cat. no. PAHS-012) according to the manufacturer's instructions. This particular PCR array profiles the expression of 84 key genes involved in programmed cell death, 5 housekeeping genes, and a set of 7 assay control primers. Raw CT values were exported from the real-time instrument software, and analyzed using the Delta Delta CT method using the RT2 Profiler™ PCR Array Data Analysis template from
Statistical analysis
To compare the RC of mutant viruses in relation to WT virus, RC values for 3 independent replication assays were calculated for WT and mutant viruses. A paired analysis with Student's t-test was performed, and p≤0.5 was considered significant.
Results
Attenuated replication of site-directed mutants K65R and M184V and recombinant HIV-1 containing multidrug resistance mutations
We compared replication capacity of various RT mutants by normalizing viruses on the basis of TCID50. We used point mutants K65R and M184V as controls in replication kinetics assays and apoptosis array analysis, because these mutations are known to be selected by key RT inhibitors, and tenofovir and FTC and have been well characterized for RC, processivity, and enzyme kinetics in vitro (4,15,17,52). Replication kinetic plots show that all the mutant viruses replicated inefficiently compared to WT (NL4-3) virus (Fig. 1A and B). Although attenuated in comparison to WT virus, PSD5.2 replicated efficiently (similarly to WT virus) in comparison to point mutants K65R and M184V. RC values were calculated by comparing antigen p24 production between WT and mutant viruses on days 7 and 10 (Table 2 and Fig. 1A). All the mutant viruses showed a significant decrease in RC in comparison to WT virus (Table 2). As the major goal of this study was to analyze AAMGs in relation to RC, we compared the differences in RCs of point mutants K65R and M184V and recombinant PSD5.1 with respect to RC of PSD5.2. A significant decrease in the RCs of K65R (0.39±0.02; p≤0.0001), M184V (0.72±0.02; p=0.0002), and PSD5.1 (0.33±0.03; p≤0.0001) was observed in comparison to PSD 5.2 recombinant virus (Fig. 1A). Since the recombinant virus PSD5.2 lacks the K65R mutation, our results suggested that K65R mutation further attenuated the replication capacity of PSD5.1 virus. A similar pattern of replication kinetics was observed when RT activity was measured; however, the production of RT activity was more pronounced in the culture supernatants obtained from infections with WT virus in comparison to mutant viruses (Fig. 1B). Since the RT assay measures the activity of RT itself, and since viruses containing altered RT are being compared with WT RT, the larger differences in replication kinetics of mutant viruses in relation to WT viruses were expected.
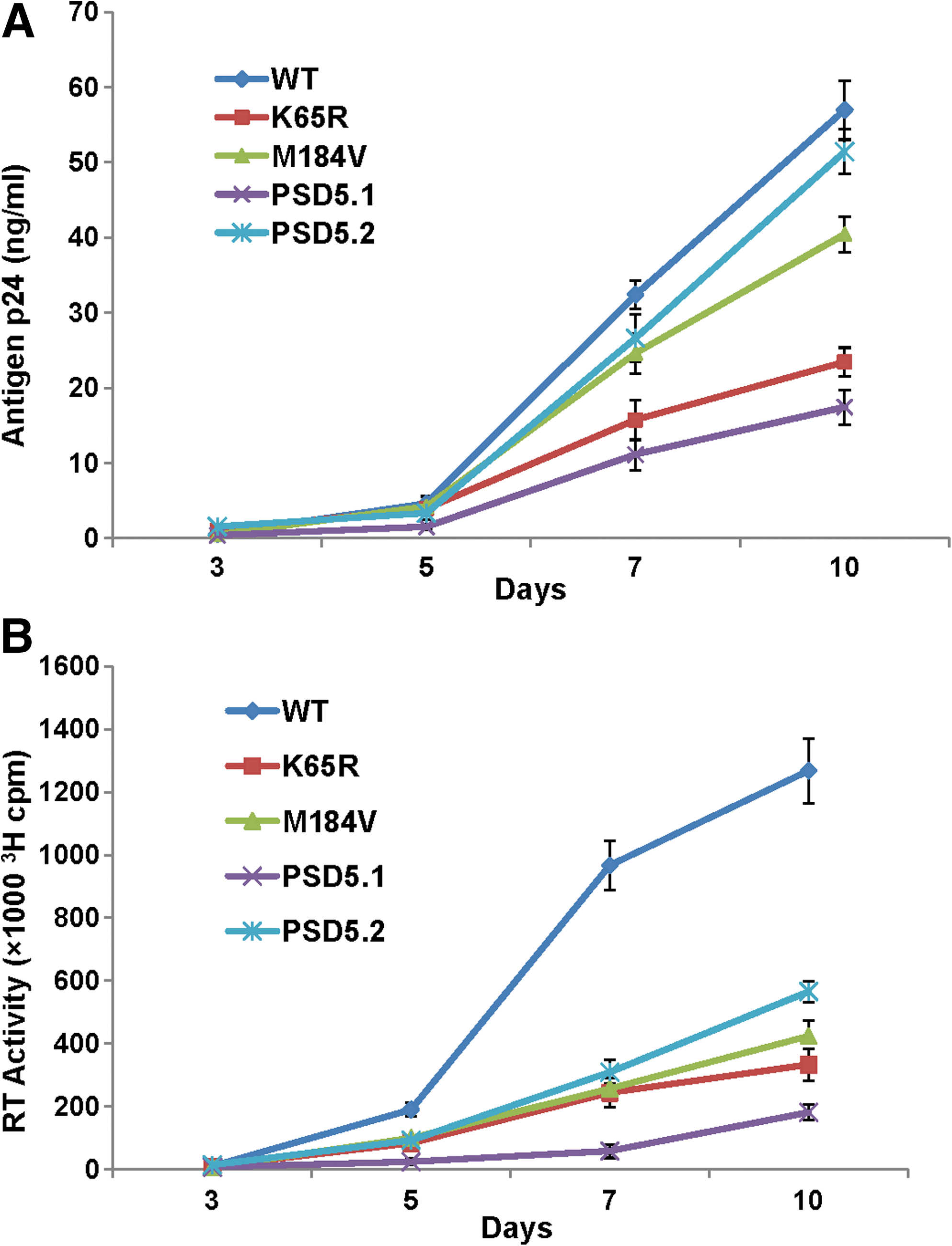
Replication kinetics of HIV RT variants. PHA-stimulated PBM cells (10×106) were infected with each virus containing 100 TCID50/1×106 PBM cells, and culture supernatants were collected at various time points to measure replication efficiencies of mutant viruses. Both HIV-1 antigen p24 (
RC values were calculated on the basis of the amount of HIV-1 antigen p24 production by mutant viruses relative to WT NL4-3. The amount of antigen p24 produced by wild-type (WT) virus was considered 100% (= 1), and fractions (mut/WT) were calculated for mutant viruses. The above RC values are obtained from three independent replication kinetics assays.
Replication independent upregulation of anti-apoptosis marker genes in PBM cells
A recent study has shown the presence of monocyte-protective signature AAMPs, even in the presence of highly replicating viruses (26). We analyzed the level of expression of 24 AAMGs in PBM cells infected with viruses with a range of RCs (Fig. 1A). We performed an RT-PCR-based apoptosis array analysis on total cellular RNA isolated from PBM cells infected with WT (NL4-3), site-directed mutant viruses containing RT mutations K65R and M184V, and recombinant viruses containing patient-derived MDR RT (PSD5.1; K65R, D67N, S68G, K70R, K103N, T215Y, and K219E) and a variant of PSD5.1 that lacks K65R (PSD5.2; D67N, S68G, K70R, K103N, T215Y, and K219E). Table 3 shows differential expression of 24 AAMGs by WT and mutant viruses in relation to the expression of AAMGs in uninfected PBM cells. As virus-induced expression of AAMGs may be delayed due to a significant decrease in RC, we compared the expression of these genes in RNA collected on day 1 and day 7 post-infection (Tables 3 and 4) with various viruses (Table 1). Out of a total of 24 marker genes tested, 11, 14, 13, 9, and 14 AAMGs were upregulated post-infection by WT, K65R, M184V, PSD5.1, and PSD5.2 viruses, respectively (Table 3). It should be emphasized that these numbers include even a minor change in the upregulation of AAMGs in relation to uninfected controls. Since the major goal of this study was to assess the differences in the expression of AAMGs in relation to RCs of viruses, we calculated the fold changes with respect to WT virus. Table 4 shows the fold change in the expression of 24 AAMGs with respect to WT virus. We found that 8 AAMGs (AKT1, BAG3, BCL2A1, BFAR, BIRC2, BNIP1, BNIP3, and CFLAR) were upregulated (>1.5 to 6-fold) with PSD5.2, 6 AAMGs (AKT1, BAG3, BCL2, BCL2L2, BNIP1, and BRAF) with K65R (>1.5 to 4-fold), 2 AAMGs (BAG1 and BNIP1) with M184V (1.6 to 2-fold), and 1 AAMG (BAG1) in PBM cells infected with PSD5.1 (1.97-fold) (Table 4). Considering that PSD5.2 viruses had higher RC compared to the other three mutant viruses (K65R, M184V, and PSD5.1), the increased expression of AAMGs appears to be interesting, and may be involved in the processes leading to increased CD4+ T-cell numbers during HAART.
PSD 5.1 (pNL4-3) contains RT fragment from a clinical isolate containing K65R, D67N, S68G, K70R, K103N, T215Y, and K219E mutations.
PSD5.2 is a variant of PSD5.1 and lacks the K65R mutation. This variant replicates efficiently in PBM cells (see Fig. 1).
Numbers in bold indicate > 1.5-fold increased expression of specific AAMG on day 7 post-infection of PBM cells by mutant viruses in comparison to WT virus.
Minus sign before numbers indicates downregulation.
PBM, peripheral blood mononuclear; AAMG, anti-apoptosis marker gene.
PSD 5.1 (pNL4-3) contains RT fragment from a clinical isolate containing K65R, D67N, S68G, K70R, K103N, T215Y, and K219E mutations.
PSD5.2 is a variant of PSD5.1 and lacks the K65R mutation. This variant replicates efficiently in PBM cells (see Fig. 1).
Numbers in bold indicate > 1.5-fold increased expression of specific anti-apoptosis marker genes observed both on day 1 as well as day 7 by mutant viruses in comparison to WT virus (transient up-regulation on day 1 only is not highlighted in bold).
Minus sign before numbers indicates downregulation.
Discussion
The current study was undertaken to develop the hypothesis that MDR HIV-induced expression of AAMGs is not dependent upon viral replication. We used site-directed mutants containing point mutations K65R and M184V, and recombinant viruses with varying RCs, and showed differential expression of AAMGs in PHA-stimulated PBM cells. Interestingly, we found that upregulation of specific AAMGs was independent of the RCs of viruses. A two- to sixfold upregulation of 8 AAMGs was observed in cells infected with PSD5.2 recombinant virus, which in fact had a significantly increased RC in comparison to K65R, M184V, and PSD5.1 recombinant viruses. On the other hand, PSD5.2 had a decreased RC in comparison to WT NL4-3 virus. These observations indicate that factors other than RC can control the regulation of specific AAMGs. We speculate that the increased and/or stable CD4+ T-cell numbers during clinical trials with HAART may be related to the upregulation of MDR virus-specific AAMPs. While the appearance of a drug resistance mutation indicates a loss of drug efficacy, there is not always a direct relationship between disease progression and the emergence of drug-resistant virus, and many fundamental questions still exist. For example, are drug-resistant mutants fully virulent and does their emergence contribute to disease progression?
While the 24 AAMGs reported in this study have been shown to be expressed in different cell types, the role of these upregulated AAMGs in the apoptosis pathway during HIV infection is not well understood. The AAMG AKT1 is a murine thymoma viral oncogene homolog 1, and is a critical mediator of growth factor-induced neuronal survival in the developing nervous system (20,23). We found about twofold upregulation of AKT1 in PBM cells infected with recombinant virus PSD5.2. Previous studies have shown that the HIV-1 envelope induces early activation of the phosphoinositide 3-kinase (PI3K)/Akt pathway in primary CD4+ T lymphocytes (24). Another study demonstrated that a transmembrane protein-tyrosine phosphatase (CD45), which is expressed on the surface of all nucleated hematopoietic cells, regulates apoptosis through upregulation of FasL and inactivation of the PI3K/Akt pathway (2). Furthermore, overexpression of WT Akt in Jurkat T cells partially protected the cells against HIV-1 envelope protein (gp120)-induced apoptosis, whereas overexpression of mutant Akt (K179M) promoted the apoptosis of these cells (2). The AAMPs BNIP1 and BNIP3 are members of the BCL-2/adenovirus E1B 19-kD-interacting protein family and are known to protect cells from virally-induced cell death. The AAMPs BCL2, BCL2A1, and BCL2L2 are members of the BCL2 protein family, a key negative regulator of cell death. Studies have shown that BCL2 protects cells in vitro and in vivo from the viral proteases, and prevents cell death following HIV infection of human lymphocytes, while reducing the yields of viral structural proteins, infectivity, and tumor necrosis factor-α (37,39,56).
Another AAMG, BFAR (bifunctional apoptosis regulator), was upregulated sixfold on day 1 and 4.83-fold on day 7 in PBM cells infected with PSD5.2. Studies have shown that BFAR protein is involved in the regulation of neuronal survival (60). A recent study has shown that BFAR, in association with an endoplasmic reticulum (ER)-associated E3 ubiquitin ligase, modulates Bax inhibitor-1 protein stability and function in ER stress (48). Interestingly, AAMG BRAF was found to be upregulated fourfold in cells infected with site-directed mutant virus containing single mutation K65R on day 7 post-infection. BRAF, a serine-threonine protein kinase, is one of the three RAF paralogs in humans. BRAF participates in the RAS-RAF-MEK-ERK pathway, a conserved protein kinase-signaling cascade that is involved in regulating a number of critical cellular functions (30,50). Recent studies have revealed that BRAF mutations are very common in malignant melanoma and are required for tumor growth and maintenance. The majority of melanoma-associated BRAF mutations involve a single point mutation, V600E, which results in greatly elevated BRAF kinase activity and constitutive activation of MAPK/ERK downstream. This resulted in BRAF-mediated partial anti-apoptotic activity (55). As stated earlier, apoptosis pathways are complex and require extensive analysis of both pro- and anti-apoptosis marker proteins and their relationship with HAART-related CD4+ T-cell protective properties.
In general, for a majority of HIV-infected patients antiretroviral therapy controls viral replication. However, in some patients drug resistance can lead to therapy failure. Nevertheless, continuation of therapy with a failing drug regimen has been shown to preserve or even lead to increases in CD4+ T-cell counts. In a recent study, investigators developed a mathematical model to study the dynamics of plasma viral RNA and CD4+ T-cell numbers and competition between fusion inhibitor enfuvirtide (ENF)-sensitive and resistant HIV during treatment interruption and re-administration. Their analysis revealed that although re-administration of ENF cannot suppress viral load, it can increase CD4+ T-cell numbers in the presence of resistant virus, which should result in clinical benefits (57). It is not known if CD4+ T-cell-protective specific anti-apoptosis marker proteins are present (or upregulated) during high viral replication. A recent study by Giri et al. (26), using monocytes from PBM cells of HIV-infected subjects, demonstrated the presence of a stable anti-apoptosis gene signature comprised of 38 genes associated with p53, CD40L, TNF, and MAPK signaling networks (26). They further showed that this gene signature is associated with cadmium chloride- or Fas ligand-induced apoptosis resistance in circulating monocytes, in contrast to increasing apoptosis in CD4 T cells from the same infected subjects. Interestingly, 28 of the 38 genes (73.7%) were stably modulated to suggest increased monocyte survival (i.e., pro-apoptotic genes were downregulated or anti-apoptotic genes were upregulated).
Another study (5) comparing apoptosis among untreated versus HAART-treated HIV-infected subjects showed that apoptosis was significantly increased in HIV-untreated subjects, but apoptosis levels did not differ between uninfected control subjects and HIV-positive subjects undergoing HAART. They further showed that despite complete suppression of viral replication, activated memory CD8+ T cells remain significantly elevated in subjects receiving HAART, suggesting the persistence of residual HIV replication. They speculated that if protease inhibitors (PIs have anti-apoptotic properties) exert a positive effect on CD4 counts beyond an antiviral effect, mechanisms other than apoptosis should be involved. It is not clear if MDR virus-induced AAMPs were involved in the protection of CD4+ T cells, leading to the stable CD4 T-cell counts seen in this study.
Our knowledge of the mechanisms involved in apoptosis resistance of monocytes and massive apoptosis of CD4+ T cells is far from complete. The diversity of strategies used by HIV-1 to manipulate the apoptotic pathway emphasizes the capacity this virus possesses to survive in its host. We have two issues. The first one is to increase the susceptibility of the infected cells, specifically monocytes/macrophages, to apoptosis, allowing reduction or elimination of the latent reservoir. The second issue is to protect uninfected by-stander CD4+ T cells by developing a sound cellular-mediated immune response against HIV-1. This could be achieved by increasing the expression of AAMPs in favor of the host. In general, apoptosis is a complex biological process in which the activation of one or more apoptotic pathways leads to a series of biochemical events, ultimately resulting in cell destruction. Various pro- and anti-apoptotic proteins are induced by both HIV-1 factors (vpr, vpu, protease, and nef), as well as host factors (cytokines/chemokines), resulting in CD4+ T-cell death (10,13,14,22,35,42,46). It is conceivable that drug-selected attenuated HIV variants will have a reduced level of apoptosis-inducing viral factors, which in turn may induce upregulation of specific anti-apoptosis marker CD4+ T-cell-protective proteins. While more comprehensive analysis is required to assess the role of AAMPs in protecting CD4+ T cells during HAART treatment, our analysis provides the interesting observation that the viruses containing MDR RT upregulate specific AAMGs in PBM cells. Future studies will assess the role of specific anti-apoptosis proteins in CD4+ T-cell apoptosis by using Si/Sh RNA technology to knock down the specific anti-apoptosis genes, and allow subsequent analysis of apoptosis in different cell types.
Footnotes
Acknowledgments
This work was supported by a VA MERIT award to P.L.S., and the U.S. Department of Veterans Affairs. We are thankful to the AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, National Institutes of Health, for providing the plasmid pNL4-3 and cell lines 293 and HeLa-CD4-LTR-β-galactosidase. This study was presented at the “Retroviruses” meeting at Cold Spring Harbor Laboratory, New York, May 24–29, 2010.
Author Disclosure Statement
No competing financial interests exist.