
Abstract
The objective of this study was to evaluate and compare the immunogenicity, safety, and tolerability of two influenza subunit vaccines, a primarily European-marketed trivalent vaccine (Agrippal®, Novartis Vaccines), and a predominantly U.S.-marketed control trivalent vaccine (Fluvirin®, Novartis Vaccines), in subjects aged 3–64 y. The immunogenicity of both vaccines was evaluated according to the Center for Biologics Evaluation and Research (CBER) criteria. This clinical trial was performed between April and December 2007 in Argentina. A total of 1893 subjects were stratified into three age groups (3–8 y, 9–17 y, and 18–64 y), and randomized in a 2:1 ratio to receive either Agrippal or Fluvirin. Adolescents and adults received one dose of vaccine intramuscularly, whereas children aged 3–8 years received two vaccine doses, administered 4 wk apart. Antibody levels were measured by means of hemagglutination inhibition assay before vaccination (baseline); 21 d after the first vaccination (adults and adolescents); and, for children aged 3–8 y, 28 d after the first vaccination and 21 d after the second vaccine dose. Adverse reactions were solicited via diary cards for 7 d after each vaccination, and unsolicited adverse events were reported throughout the study period. Both vaccines were safe and well-tolerated, and elicited robust immunogenic responses in all age groups, meeting both CBER licensure criteria for all three viral strains after completion of the age-recommended vaccination schedule. These findings support the use of the trivalent subunit influenza vaccines Agrippal and Fluvirin for universal vaccination campaigns on an annual basis.
Introduction
Because of the enormous annual burden of influenza resulting from hospitalization costs, lost productivity from missed work days, and lost lives (17), the Centers for Disease Control and Prevention's (CDC) Advisory Committee on Immunization Practices (ACIP) has expanded the recommendations for influenza vaccination to include adults 50 y of age and older (2000) (3), healthy children aged 6–23 mo of age, women who would be pregnant during the influenza season (2004) (14), persons with conditions that compromise respiratory function or handling of respiratory secretions (2005) (15), and all children 24–59 mo of age and their household contacts and out-of-home caregivers (2006) (28). In February 2008, the ACIP voted to further expand the recommendations for influenza vaccination to include children 5–18 y of age as soon as feasible, but no later than 2009/2010 (9). Current recommendations include annual vaccination for all persons aged ≥6 mo (10,25). Increased demand for influenza vaccines resulting from broader recommendations, along with distribution delays, have resulted in vaccine supply shortages in the U.S. over the past influenza seasons (27).
Agrippal® (Novartis Vaccines and Diagnostics, Siena, Italy) is a highly-purified subunit, inactivated, egg-based trivalent influenza vaccine. It is propagated in eggs and inactivated by formaldehyde. In addition, antibiotics (kanamycin and neomycin sulfate), polysorbate 80, and cetyltrimethylammonium bromide, are used during the manufacturing process, and trace amounts may therefore be present in the final vaccine. It was initially licensed in Italy in 1986, where it has been recommended for prophylaxis of influenza in subjects aged 6 mo and older, especially those who are at an increased risk of associated complications. Agrippal is now licensed for the same indication in 61 countries worldwide. Cumulatively from the first launch through April 30, 2011, the estimated subject exposure was approximately 115 million for prophylaxis of influenza in toddlers, children, adolescents, adults, and the elderly (21). Subunit vaccines are known to be well-tolerated and safe because they do not contain nucleoprotein or matrix proteins, they contain low levels of DNA contaminants, and they show a higher degree of purity compared with other types of vaccines (4,29,32). In the present article we report the results of a Phase III trial designed to assess the immunogenicity, safety, and tolerability of Agrippal compared with Fluvirin® (Novartis Vaccines and Diagnostics, Liverpool, U.K.) in healthy subjects 3–64 y of age. Immunogenicity of both vaccines was evaluated according to the U.S. Food and Drug Administration/Center for Biologics Evaluation and Research criteria (CBER) (33). Fluvirin is an inactivated, subunit, conventional egg-derived, trivalent influenza vaccine containing either thimerosal as a preservative (multidose vials), or trace amounts of thimerosal (pre-filled syringes). It is approved for subjects 4 y of age or older, and is well-established in the U.S. market, with a confirmed safety profile (12,29). The manufacture of Fluvirin involves growing of virus in eggs, and subsequent processing with neomycin, polymyxin, beta-propiolactone, nonoxynol 9, and formaldehyde.
Materials and Methods
Study design, subjects, and vaccines used
This Phase III, observer-blind, randomized controlled multicenter study was carried out between April and December 2007 in Argentina. The study was undertaken in compliance with Good Clinical Practice guidelines and the Declaration of Helsinki, and the study protocol was approved by the ethics committees of all the participating centers. Before enrollment, written informed consent was obtained from subjects, parents, or legal guardians.
The primary study objective was to evaluate immunogenicity after one vaccination of Agrippal in healthy adults aged 18–64 y according to the CBER Guidance for Industry issued in May 2007 (33). Secondary objectives were to evaluate the immunogenicity of Agrippal in children and adolescents, and Fluvirin according to CBER criteria. The purpose of the control arm (Fluvirin), was primarily to provide a comparative assessment for safety, not immunogenicity or effectiveness. Safety and tolerability objectives included the evaluation of solicited local and systemic reactogenicity, and spontaneously reported adverse events (AEs) and serious AEs (SAEs).
Participants were 3–64 y of age, and were in good health as determined by medical history, physical examination, and clinical judgment of the investigator. Main exclusion criteria included current infectious or serious disease, a history of hypersensitivity to study vaccine components or eggs, impaired or altered immune function, laboratory-confirmed influenza, and vaccination against influenza within 6 mo before enrollment. In addition, children 3–8 y of age who had previously received an influenza vaccine were excluded from the study.
Each study vaccine dose [Agrippal (eTIV_a), and Fluvirin (eTIV_f)] contained 15 μg of viral hemagglutinin for each of the virus strains recommended for the 2007 Southern Hemisphere influenza season: A/New Caledonia/20/99 (H1N1)-like, A/Wisconsin/67/2005 (H3N2)-like, and B/Malaysia/2506/2004-like.
At enrollment, participants were stratified according to age and were randomized in a 2:1 ratio to receive either eTIV_a or eTIV_f. Vaccines were provided as pre-filled syringes of 0.5 mL per injection (eTIV_a), or multi-vial doses of 0.5 mL per injection (eTIV_f). Randomization assignments were supplied to designated unblinded study staff in sealed envelopes. Adolescents (9–17 y), and adults (18–64 y), received one dose of vaccine, whereas children aged 3–8 y received two doses of vaccine administered 4 wk apart. Vaccines were administered intramuscularly, preferably in the deltoid muscle of the nondominant arm.
Blood samples for immunogenicity analyses were collected before vaccination (day 1, visit 1) for all age groups. For adolescents and adults, blood samples were collected again at 21 d after vaccination (day 22, visit 2). In children aged 3–8 y, blood samples were collected at 28 d after the first vaccination (day 29, visit 2), and at 21 d after the second vaccine dose (day 50, visit 3). Solicited local and systemic adverse reactions were recorded on a diary card for 7 consecutive days following each vaccination. Reports of all unsolicited AEs and SAEs were collected for the 22-day (age groups 9–17 y and 18–64 y), or 50-day study period (children aged 3–8 y). After the last scheduled clinical visit, the subjects were followed for safety via telephone contact at 6 mo after the last study vaccination to collect SAEs, AEs necessitating a physician's visit, and AEs leading to study withdrawal.
Immunogenicity assessment
Blood samples taken for immunologic assays were centrifuged, and sera were stored at −18°C or below until shipped to the Novartis Vaccines Clinical Serology Laboratory in Marburg, Germany, for further analysis. Antibodies against the vaccine strains of A/H1N1, A/H3N2, and B were measured using hemagglutination inhibition (HI) assay, according to standard methods (22).
HI antibody responses were expressed as geometric mean titer (GMT), and geometric mean ratio, seroprotection rates were defined as the percentage of subjects with HI titers ≥40, and seroconversion rates were defined as the percentage of subjects achieving at least a fourfold increase in HI titer from a seropositive pre-vaccination titer (≥1:10), or a rise from <1:10 to ≥1:40 in those who were originally seronegative.
Safety and tolerability assessment
Subjects, or subjects' legal representatives, were provided with diary cards and asked to record the occurrence of any adverse local or systemic reactions for 7 d following vaccination. Vaccinees were observed for at least 30 min after each vaccination to monitor for immediate adverse reactions. Solicited local reactions included ecchymosis, erythema, induration, swelling, and pain. Systemic solicited reactions were chills, malaise, myalgia, arthralgia, headache, sweating, fatigue, and fever. Other indicators of reactogenicity were remaining at home due to vaccine reactions, and the use of analgesics/antipyretic medications for vaccination-induced fever. Severe solicited reactions were defined as those that were either injection-site reactions >100 mm diameter, reactions leading to an inability to perform normal daily activities, or axillary temperature ≥40°C.
Spontaneously reported AEs were collected throughout the study period and assessed for severity, seriousness, and relationship to the study vaccine according to the opinion of the investigator. Any SAE was to be reported to the study sponsor within 24 h of awareness.
Statistical analyses
Statistical analyses were performed using SAS® version 9.1. Sample size calculation was based on the assumption that at study day 22, the true percentages of subjects with seroprotection (HI>1:40) was 80%, and the percentage of subjects with seroconversion was 50%. It was planned to have 1620 evaluable subjects randomized in a 2:1 ratio to receive either Agrippal (360 subjects in each age stratum), or Fluvirin (180 subjects in each age stratum). Allowing for a 10% dropout rate, at least 1800 study subjects were to be enrolled in this trial. All analyses were run by age stratum to evaluate the immunogenicity of the vaccine groups in compliance with the immunogenicity criteria stated in the CBER Guidance document issued in May 2007.
The precision of the estimates for the investigational vaccine within each age stratum (Agrippal; n=360), were expressed by the 95% Clopper-Pearson CI, for several possible observed values of the seroprotection rate and of the seroconversion rate. For the primary immunogenicity objective, with 360 evaluable subjects in each age stratum, the lower limits of the 95% CI around the estimated percentage of subjects achieving seroprotection (HI titer ≥1:40), or with seroconversion were considered to meet or exceed the threshold level of 70% and 40%, respectively, if seroprotection was at least 75% (95% CI 70.2%,79.4%), and the percentage of subjects with seroconversion was at least 46% (95% CI 40.8%,51.3%).
The GMTs, their ratios, and proportions, reflecting the immunogenicity endpoints and the corresponding two-sided 95% CIs, were calculated within each age and vaccine group. Although immunogenicity was assessed for both vaccines, the main purpose of the control vaccine eTIV_f was to provide a comparative assessment for safety. Safety and tolerability data were summarized by age and vaccine group, providing the percentage of subjects reporting an event.
Results
Population
Subject disposition is illustrated in Fig. 1. Altogether, 1893 subjects were enrolled in the study (692 adults age 18–64 y, 600 children and adolescents age 9–17 y, and 601 children age 3–8 y), of which 67% were vaccinated with eTIV_a, and 33% received eTIV_f. Overall, 98% (eTIV_a), and 97% (eTIV_f), of subjects completed the study. Premature withdrawals included lost to follow-up (2%), and withdrawal of consent (<1%). Baseline characteristics of enrolled subjects per age strata and vaccine group are presented in Table 1, which shows that gender, race, height, weight, and prior influenza vaccine were evenly distributed across vaccine groups. One subject aged 18 years (Fluvirin group) was mistakenly enrolled in the adolescent age strata but was analyzed for safety in the adult age strata. As defined by the protocol, all children aged 3–8 y were influenza vaccine-naive.

Study outline (eTIV_a, egg-derived seasonal trivalent subunit influenza vaccine Agrippal; eTIV_f, egg-derived seasonal trivalent subunit influenza vaccine Fluvirin).
Numbers are presented as proportion or as mean±standard deviation.
eTIV_a, egg-derived seasonal trivalent subunit influenza vaccine Agrippal; eTIV_f, egg-derived seasonal trivalent subunit influenza vaccine Fluvirin.
Immunogenicity
Overall, 1893 subjects were vaccinated and provided at least one blood draw (i.e., modified intent-to-treat [MITT] population), and were therefore included in the MITT population (defined as all randomized subjects in the enrolled population who received a study vaccination and provided at least one evaluable serum sample). A total of 1262 subjects received at least one vaccination of eTIV_a, and 631 subjects were vaccinated with eTIV_f. Pre-vaccination evaluation of seroprotection rates, and post-vaccination evaluation of seroprotection and seroconversion rates according to CBER across age and vaccine groups and virus strains are presented in Table 2 (adults and adolescent populations), and Table 3 (children aged 3–8 y). For each influenza virus strain, pre-vaccination seroprotection rates were similar between vaccine groups within each age stratum, with higher rates observed against the A strains than the B strain, and in subjects aged 9–17 y. At day 22, 3 wk after vaccination with eTIV_a, strong immune responses were evident in the adult population, and both CBER criteria were met for all virus strains. At 3 wk after vaccination, over 90% of adults who received eTIV_a exhibited seroprotective HI titers (HI≥40) against the A/H1N1, A/H3N2, and B strains (range of the lower bounds of the 95% CI 88%,94%; Table 2), and the proportion of subjects achieving seroconversion for the A and B strains exceeded the 40% threshold (range of the lower bounds of the 95% CI 68%,73%; Table 2). Similar results were observed for adults that received eTIV_f. Three weeks after vaccination, a large proportion of subjects exhibited seroprotective titers, and showed seroconversion against the A/H1N1, A/H3N2, and B strains, with lower bounds of the two-sided 95% CI exceeding the required thresholds (Table 2).
eTIV_a, egg-derived seasonal trivalent subunit influenza vaccine Agrippal; eTIV_f, egg-derived seasonal trivalent subunit influenza vaccine Fluvirin; GMT, geometric mean titer; GMR, ratio of day 22/day 1 geometric mean HI titers.
Seroprotection is defined as HI titer ≥40, seroconversion is defined as negative pre-vaccination serum (ie, HI titer <10),and post-accination HI titer ≥40, or significant increase defined as at least a fourfold increase from nonnegative (≥10) pre-vaccination HI titer; CBER criteria: the lower bound of the two-sided 95% CI should (1) meet or exceed 40% in subjects achieving seroconversion on HI assay, and (2) meet or exceed 70% in subjects achieving seroprotection (HI antibody titer ≥40). Bold type indicates that the CBER criteria were met.
eTIV_a, egg-derived seasonal trivalent subunit influenza vaccine Agrippal; eTIV_f, egg-derived seasonal trivalent subunit influenza vaccine Fluvirin; GMT, geometric mean titer; GMR, ratio of day 22/day 1 geometric mean HI titers.
Seroprotection is defined as HI titer ≥40, seroconversion is defined as negative pre-vaccination serum (ie, HI titer <10), and post-vaccination HI titer ≥40, or significant increase defined as at least a fourfold increase from nonnegative (≥10) pre-vaccination HI titer. CBER criteria: the lower bound of the two-sided 95% CI should (1) meet or exceed 40% in subjects achieving seroconversion on HI assay; and (2) meet or exceed 70% in subjects achieving seroprotection (HI antibody titer ≥40). Bold type indicates CBER criteria were met.
In adolescents aged 9–17 y, similar trends were observed, and CBER criteria for seroprotection and seroconversion were met for both eTIV_a and eTIV_f at 3 wk after vaccine administration (Table 2).
Children 3–8 y of age received two vaccinations administered 4 wk apart. For both vaccine groups, CBER criteria for seroprotection were met against the AH1N1 and A/H3N2 strains, and criteria for seroconversion were fulfilled against all three influenza strains, even after the first vaccine dose (Table 3). At day 50, 3 wk after the second vaccination, both CBER criteria were met for all strains for both vaccine groups.
Safety and tolerability
All enrolled (n=1893) subjects were included in the data set for the safety and tolerability analyses. The proportion of subjects reporting solicited local and systemic adverse reactions, within 7 d post-vaccination were balanced between the eTIV_a and eTIV_f groups, and was slightly higher in the adolescent and adult groups compared with children aged 3–8 y (Tables 4 and 5). In this youngest age group receiving two doses of vaccine, the frequency and severity of solicited reactions did not increase after the second dose was administered. The majority of solicited reactions were mild to moderate and resolved within days. Local reactions were reported in 34% (eTIV_a) and 31% (eTIV_f), and 35% (eTIV_a) and 38% (eTIV_f), in the adolescent and adult populations, respectively. Systemic reactions were reported in 23% (eTIV_a) and 25% (eTIV_f), and 32% (eTIV_a) and 36% (eTIV_f), in adolescents and adults, respectively. In the age cohort 3–8 y, local reactions were reported in 23% (eTIV_a) and 28% (eTIV_f) of children after the first vaccination, and in 17% (eTIV_a) and 20% (eTIV_f) after the second vaccine dose. Systemic reactions were reported in 16% (eTIV_a) and 19% (eTIV_f), and 10% (eTIV_a) and 11% (eTIV_f) of subjects, after the first and second vaccination, respectively. The most commonly reported local reaction was pain at the injection site in adults and adolescents (Table 4), and children (Table 5). The most frequently reported systemic reactions across the age cohorts was headache (Table 4 for adults/adolescents, and Table 5 for children). Fever≥38°C was reported in only <1–3% across age and vaccine groups, with no reports of severe fever in subjects aged 3–8 y and 9–17 y, and <1% in adults (eTIV_a). Severe local (pain only), and systemic solicited reactions, were reported rarely, with 2% (headache in adults), and only ≤1% of any other reaction classified as severe, equally divided across vaccine groups.
eTIV_a, egg-derived seasonal trivalent subunit influenza vaccine Agrippal; eTIV_f, egg-derived seasonal trivalent subunit influenza vaccine Fluvirin.
eTIV_a, egg-derived seasonal trivalent subunit influenza vaccine Agrippal; eTIV_f, egg-derived seasonal trivalent subunit influenza vaccine Fluvirin.
Overall, the percentages of spontaneously-reported AEs during the post-vaccination periods (22 d [adults and adolescents], 50 d [children 3–8 y], and 6-mo follow-up) were balanced between vaccine groups (Fig. 2A and B). Possibly related AEs were uncommon (2% in adults, 1% in adolescents, and ≤1% in children), and were mainly typical post-vaccination reactions; all were mild to moderate in severity and resolved before study termination. The most commonly experienced unsolicited AEs in adults were nasopharyngitis (2–3%), influenza-like illness, and headache (1–2%). In 9- to 17-year-olds, nasopharyngitis (2–3%), rhinitis (<1–2%), pharyngitis and gastroenteritis (<1–1%) were most common. In the youngest age cohort, bronchitis, nasopharyngitis, pharyngitis, and vomiting (all less than 2%) were most frequently reported. During the 6-month follow-up period, unsolicited AEs were reported in 4–6%, 1%, and 1–2% of adults, adolescents, and children, respectively.
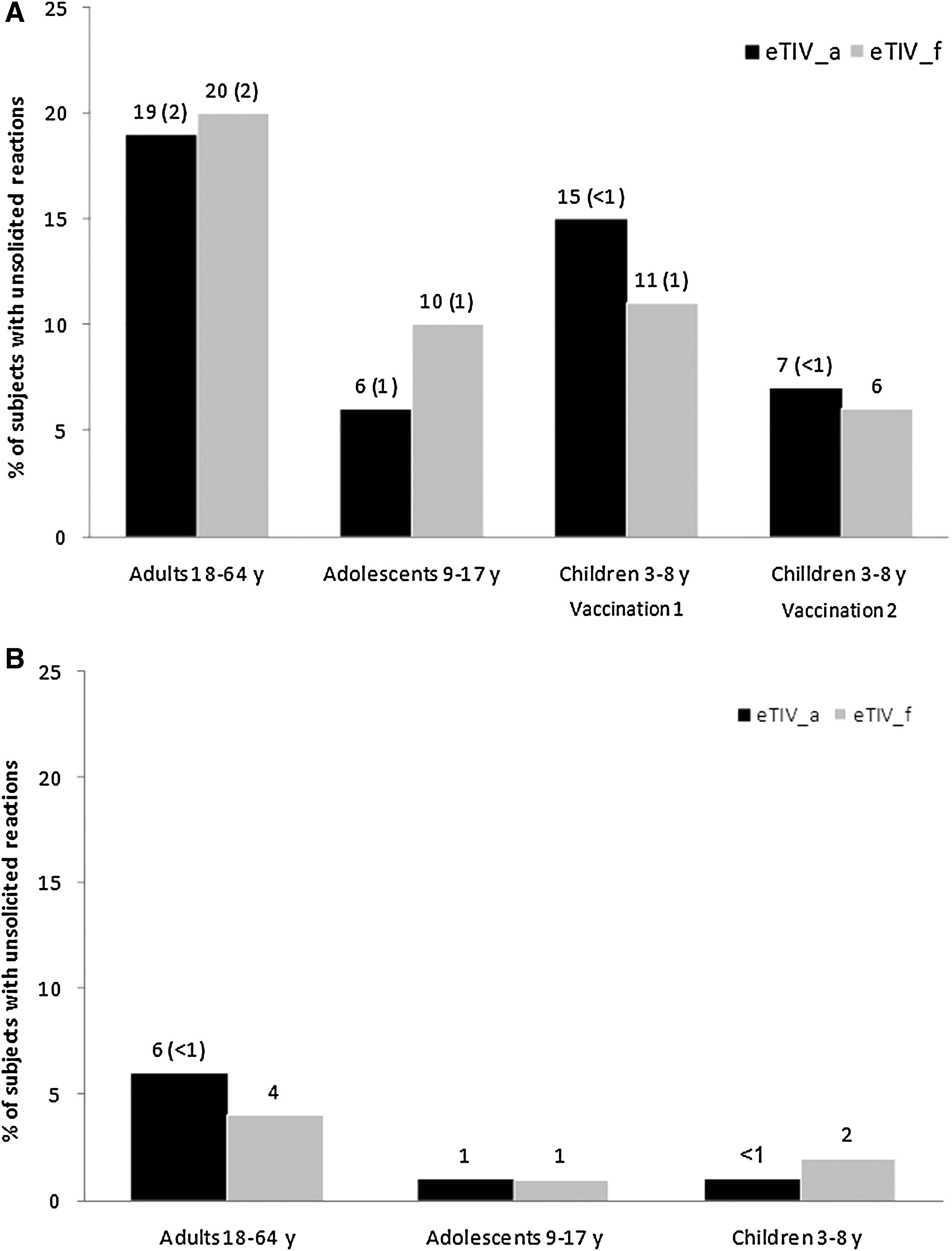
Proportion of subjects (%) reporting unsolicited adverse events (and in parentheses % of at least possibly related adverse events) 22 days for adults and adolescents and 50 days for children postvaccination (
Throughout the study period, a total of 18 SAEs were reported; 17 SAEs were considered not related to the vaccine, and 1 SAE was considered to be possibly vaccine related. Three SAEs were reported during the post-vaccination period (9–17 y: 1 [eTIV_a], 3–8 y: 2 [eTIV_a]), and 15 SAEs were reported during the 6-month follow-up period (adults: 8 [eTIV_a: 6, eTIV_f: 2 ]; 9–17 y: 3 [eTIV_a: 3]; and 3–8 y: 4 [eTIV_a: 1, eTIV_f: 3]). The possibly-related SAE (spontaneous abortion, eTIV_a) occurred in a 26-year-old woman approximately 2 mo after vaccine injection. There were no deaths or AEs leading to study withdrawal. Altogether, the safety and tolerability profiles in the age cohorts 18–64 y, 9–17 y, and 3–8 y, were comparable between eTIV_a and eTIV_f.
Discussion
This is the first study to evaluate the safety and immunogenicity of a primarily European-marketed vaccine, and a predominantly U.S.-marketed vaccine, in a single study in a large healthy population aged 3–64 y.
Results of this study show that both vaccines elicited robust immunogenic responses in adult and adolescent age cohorts, and by day 22 after vaccination, both CBER criteria for licensure of influenza vaccines were met for all three virus strains. In children aged 3–8 y, CBER criterion for seroconversion was achieved in both vaccine groups against all vaccine strains, even after first vaccine dose, whereas the criterion required for seroprotection was met against both A influenza strains only, after first vaccine dose, and against all strains after completion of the vaccination schedule. Across the age strata a single vaccination with eTIV_a or eTIV_f induced between 9- and 36-fold increases in HI titer, with further increases following the second vaccination in children. It should be noted that baseline seroprotection rates were lower for the B than the A strains, which may be explained by the lower sensitivity of the HI assay against the B strain (18). The high baseline titers seen across the age groups, and especially against the A/H3N2 strain in the younger populations, could be due to the fact that the influenza season started during the enrollment period of the study.
The results from this study showing that both Agrippal and Fluvirin were well-tolerated and severe reactions were infrequent in all age groups are consistent with previously published results of systematic reviews and meta-analyses of randomized clinical trials, showing that subunit vaccines are safe and well-tolerated (1,2,4,7,13,23,29,35). For example, a recent systematic review and meta-analysis reported the results from 33 influenza vaccine studies including subunit vaccines (1). It was observed, using data from 8462 vaccinees, that the vaccine reactions were mild and transient, and there was an absence of serious vaccine-related adverse events (1). The equally low reactogenicity of both eTIV_a and eTIV_f is consistent with the higher purity of subunit vaccines compared to split vaccines (4,6,29). The good tolerability profile of both vaccines is also supported by the extensive post-marketing database since licensure (unpublished data). All AEs reported in the trial were either common illnesses to be expected in the enrolled population, or known AEs associated with influenza vaccination (20). Although a spontaneous abortion in a 26-year-old woman that occurred 60 d after injection was judged as possibly related to vaccination, there is no evidence from pre-clinical and clinical trials or from post-marketing surveillance that influenza vaccination can cause fetal damage or spontaneous abortion.
This large clinical trial of 1893 participants aged from 3–64 y, confirms the safety and immunogenicity of eTIV_a and eTIV_f in the pediatric, adolescent, and adult populations. The purity of subunit vaccines explains their favorable tolerability profile, and supports the use of Agrippal and Fluvirin for vaccinating the general population on an annual basis, along with those at high risk such as children, the elderly, and those with underlying chronic diseases (2,7,23). Although many individuals in this population were not at increased risk of complications from influenza, vaccination of healthy individuals slows down or prevents transmission of influenza, reduces rates of influenza-like illness and associated complications, and can yield other significant health and economic benefits, such as reduced work absenteeism, as well as cost benefits (5,19). Therefore, an increased demand for influenza vaccines could be met by highly purified inactivated subunit vaccines such as Agrippal and Fluvirin, which have a well-established safety and tolerability profile, and a large post-marketing database.
Footnotes
Acknowledgments
Novartis Vaccines supported the costs of the study, supplied purified influenza virus antigens, and participated in the analysis of the clinical data. We are grateful to all the medical doctors and staff in the medical center and laboratories for their collaboration. We also thank Luiz Jacintho da Silva, M.D., and Mike Penlington, Ph.D., for their critical review of the manuscript, and Indah Andi Lolo and Patricia de Groot, Ph.D. (CHC Europe) for assistance in preparing the manuscript.
Author Disclosure Statement
Dr. Daniel Stamboulian has received speaker honoraria and funding for medical education activities from Novartis. Elena Fragapane, Daniela Casula, Michele Pellegrini, and Nicola Groth are Novartis employees. Dr. Miguel Tregnaghi, Dr. Paula Carina Vanadía, Dr. Jorge Pablo Tregnaghi, and Dr. Miriam Calvari have no conflicts of interest to declare.