
Abstract
The innate immune response induced by Hantavirus is responsible for endothelial cell dysfunction and viral pathogenicity. Recent studies demonstrate that TLR4 expression is upregulated and mediates the secretion of several cytokines in Hantaan virus (HTNV)-infected endothelial cells. To examine viral interactions with host endothelial cells and characterize the innate antiviral responses associated with Toll-like receptors, we selected TLR4 as the target molecule to investigate anti-hantavirus immunity. TLR4 mRNA-silenced EVC-304 (EVC-304 TLR4–) cells and EVC-304 cells were used to investigate signaling molecules downstream of TLR4. The expression of the adaptor protein TRIF was higher in HTNV-infected EVC-304 cells than in EVC-304 TLR4– cells. However, there was no apparent difference in the expression of MyD88 in either cell line. The transcription factors for NF-κB and IRF-3 were translocated from the cytoplasm into the nucleus in HTNV-infected EVC-304 cells, but not in HTNV-infected EVC-304 TLR4– cells. Our results demonstrate that TLR4 may play an important role in the antiviral immunity of the host against HTNV infection through an MyD88-independent signaling pathway.
Introduction
Hantavirus is a noncytopathogenic virus that targets primarily vascular endothelial cells. Therefore it has been suggested that hantavirus pathogenesis is linked to the cellular immune response, which is activated in infected vascular endothelial cells. The importance of the immune response for hantavirus pathogenesis, especially the innate immune response, is widely recognized. Among the many different receptors that participate in the recognition of microbial invaders, Toll-like receptors (TLRs) play important roles in mediating the innate immune response (2). Because TLRs are involved in the pathogenesis of viruses, such as the respiratory syncytial virus (RSV), retrovirus, coxsackievirus, and mouse mammary tumor virus (MMTV) (8,18,32,37), we investigated whether this family of receptors participates in the course of HFRS pathogenesis. We previously demonstrated that the expression of TLR4 is upregulated in HTNV-infected human vascular endothelial cells (EVC-304 cells), and mediate the upregulated expression of TNF-α, IFN-β, and IL-6 in HFRS (10). However, the role of TLR4 in HTNV infection is still unknown.
Evidence suggests that several pathogen-associated molecular patterns (PAMPs) upregulate the expression of TLR4. These molecules include lipopolysaccharide (LPS) from gram-negative bacteria, the envelope protein from MMTV, and the fusion protein from RSV (18,32). Moreover, endogenous molecules, such as heat-shock proteins, hyaluronic acid and β-defensin 2, interact with TLR4 (5,28,36). TLR4 signaling is mediated by MyD88-dependent and MyD88-independent (TRIF-dependent) pathways (23). Following recognition by PAMPs, TLR4 undergoes oligomerization and recruits downstream adaptors through interactions with the TIR (Toll-interleukin-1 receptor) domains. TIR domains contain three highly conserved regions that mediate protein–protein interactions between the TLRs and signal transduction adaptor proteins. The TIR domain of TLR4 is critical for signal transduction; a single point mutation in the TIR domain can abolish the response (30). There are five TIR domain-containing adaptor proteins: MyD88 (myeloid differentiation primary response gene 88), TIRAP (TIR domain-containing adaptor protein, also known as Mal; MyD88-adapter-like), TRIF (TIR domain-containing adaptor that induces IFN-β expression), TRAM (TRIF-related adaptor molecule), and SARM (sterile α and HEAT-Armadillo motifs-containing protein) (23,27). MyD88 was first described as a myeloid differentiation primary response gene (22), a critical adaptor in the interleukin-1 receptor (IL-1R) signaling pathway (1,25). TIRAP facilitates the association between MyD88 and the TLR4 cytoplasmic domain to initiate MyD88-dependent downstream signaling (11). Studies with knockout mice indicate that TRIF and TRAM mediate MyD88-independent signaling (9,40,41). However, the role of SARM in vivo is unclear. The transcription factors NF-κB and IRF-3 are downstream molecules in the TLR4 signaling pathway. Once TLR4 is activated, NF-κB and IRF-3 translocate from the cytoplasm into the nucleus and mediate the activation of proinflammatory cytokines and chemokines as well as type I interferon genes (23).
In this study, we investigated the signaling pathway of TLR4 in HTNV-infected EVC-304 cells. TLR4-silenced EVC-304 cells indicate that MyD88-independent signaling likely mediates the TLR4 signal, and that TLR4 may play an important role in the pathogenesis of HTNV.
Materials and Methods
Reagents, virus, and cell lines
EVC-304 and EVC-304 TLR4– cells were provided by Dr. Jiang (10). In EVC-304 TLR4– cells, TLR4 mRNA is specifically silenced by siRNA. Both cell lines were maintained in RPMI 1640 (Hyclone, Logan, UT) supplemented with 10% fetal bovine serum (Gibco, Invitrogen, Grand Island, NY), 100 IU penicillin, and 100 μg/mL streptomycin.
HTNV strain 76–118 (19) was proliferated in Vero E6 cells, purified with Viraffinity™ 50 (Biotech Support Group, North Brunswick, NJ) and treated with polymyxin B.
LPS (InvivoGen, San Diego, CA) was used as a positive control for HTNV stimulation. Complete RPMI 1640 was used as a mock-infected control.
Stimulation and infection
For the stimulation experiments, EVC-304 cells were incubated overnight in culture medium on Costar culture plates (Cambridge, MA). Following the overnight incubation, the cells were stimulated by incubation in RPMI-1640 containing LPS (1 μg/mL), or were infected with HTNV 76–118 at a multiplicity of infection (MOI) of 5. The virus was allowed to adsorb at 37°C in 5% CO2 for 2 h. The cells were then washed, and the medium was replaced with fresh RPMI 1640 containing 10% FBS.
Semiquantitative RT-PCR analysis for the detection of TLR4 in EVC 304 TLR4– cells
Total RNA was extracted from cells with TRIzol reagent (Invitrogen Life Technologies, Carlsbad, CA). Briefly, 1 mL TRIzol reagent was used to lyse 1×106 cells. Next, 200 μL chloroform and 500 μL 2-propanol were added to separate the RNA from DNA and proteins. Finally, after washing with 75% ethanol, the RNAs were dried and dissolved in 30 μL of DEPC-treated water. The RNA was quantitated with a RiboGreen Kit (Molecular Probes, Eugene, OR). The ReverAid™ First Strand cDNA Synthesis Kit (Fermentas, Vilnius, Lithuania) was used to synthesize cDNA from the total RNA according to the manufacturer's protocol.
TLR4 was amplified by PCR with Taq polymerase (Fermentas, Vilnius, Lithuania) with 30 cycles at 94°C for 40 sec, 56°C for 40 sec, and 72°C for 40 sec. β-Actin was used as an internal control for the RT-PCR experiments. The primers were synthesized by Invitrogen. The primer sequences were as follows: TLR4 (NM138554): forward: 5′-AGAAAACAGAAAGAGACATTG-3′, reverse: 5′-TCAAACATCACTTGTTCTGAA-3′; β-Actin (NM001101): forward: 5′-GGCTACAGCTTCACCACCAC-3′, reverse: 5′-GCACTGTGTTGGCGTACAGG-3′.
RT-PCR was performed with a MyCycler thermal cycler (Bio-Rad, Hercules, CA). The RT-PCR products were separated by gel electrophoresis on 2.0% agarose gels containing 0.5 μg/mL ethidium bromide (EB) in Tris-borate ethylenediaminetetraacetic acid (TBE) buffer, visualized with a Vilber Lourmet transilluminator and photographed with BiocaptMW Version 10.02 software for Windows (Vilber Lourmat, Cedex, France).
Quantitative real-time PCR analysis
Total RNA was extracted and cDNA was synthesized as described above. Real-time PCR was performed with the LightCycler system (Roche Diagnostics GmbH, Mannheim, Germany). The sequences of the oligonucleotides (synthesized by Invitrogen) were as follows: MyD88 (NM002468): Forward: 5′-TCAAGGGTAGAGGTGGGCAC-3′, Reverse: 5′-CGGCTTTCGCTTTCCGAGAA-3′; Taqman probe: 5′(FAM)-CGCCGCCCTGCCCTACAATCTGG(Eclipse)-3′; TRIF (NM182919): Forward: 5′-AGACTGTGTCATCCCCTTCCT-3′, Reverse: 5′- CCACCTTCCTGGCGAAGATC-3′; Taqman probe: 5′(FAM)-TGTTCGTCCAGCCGCACCAGCC(Eclipse)-3′; GAPDH (NM002046): Forward: 5′- CTTAGCACCCCTGGCCAAG-3′, Reverse: 5′-GATGTTCTGGAGAGCCCCG-3′; and Taqman probe: 5′(FAM)-CATGCCATCACTGCCACCCAGAAGA(Eclipse)-3′.
All sample measurements were performed at least in triplicate, and the relative expression of MyD88 and TRIF was determined by normalizing to GAPDH expression to calculate the fold change in value. Gene expression was calculated with the ΔΔCt method (20). Each sample was assayed for the number of PCR cycles required to cross the threshold of the linear range of the reaction (Ct). The amplification efficiencies of the target gene and the reference gene were approximately equal.
Western blotting
Cell extracts were prepared as previously described (33). Subsequently, 10 μg of total protein from each sample was added to Laemmli loading buffer, boiled for 5 min, resolved by 10% SDS-PAGE, and electroblotted onto nitrocellulose membranes (Hybond C, Amersham Biosciences, Little Chalfont, Buckinghamshire, U.K.). After blocking for 1 h in TBST containing 5% non-fat milk, the membranes were washed three times in TBST and probed with primary antibodies [(mouse polyclonal antibody against human MyD88; Biodesign, Carmel, NY), (rabbit monoclonal antibody against human TRIF; Chemicon, Millipore, Billerica, MA), and (mouse monoclonal antibody against human β-actin; Zhongshanjinqiao Biotech, Beijing, China)] at 4°C overnight. The membranes were incubated with horseradish peroxidase (HRP)-conjugated Cruz Marker™-compatible secondary antibodies [(goat anti-mouse IgG-HRP; Biodesign) and (goat anti-rabbit IgG-HRP; Chemicon, Millipore)], and visualized with enhanced chemiluminescence (Western Blot Luminol Reagent; Santa Cruz Biotechnology, Santa Cruz, CA), in accordance with the manufacturer's instructions. The housekeeping gene β-actin was used as an internal control.
Immunofluorescence assays
Immunofluorescence assays (IFA) were performed as previously described (39). After incubation with mouse monoclonal antibody against human NF-κB (dilution 1:200; Abcam, Cambridge, U.K.), and IRF-3 (dilution 1:200; Abcam, Cambridge, U.K.) for 2 h at 37°C, the EVC-304 cells were washed three times with PBS for 5 min per wash. Next the cells were incubated with fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG (dilution 1:500; SouthernBiotech, Birmingham, AL) for 1 h at 37°C. The nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; Roche Diagnostics GmbH, Mannheim, Germany). An Olympus BX51 fluorescence microscope (Olympus, Tokyo, Japan) with the appropriate fluorescence filters was used to capture images.
Electrophoretic mobility shift assay (EMSA)
The nuclear extracts were prepared from the EVC-304 and EVC-304 TLR4– cells with a nuclear protein extraction kit (Viagene Biotech, Ningbo, China). EMSA was performed with a non-radioactive EMSA kit in accordance with the manufacturer's instructions (Pierce, Rockford, IL). The sequences of the oligonucleotides used were as follows: 5′-AGTTGAGGGGACTTTCCCAGGC-3′ for NF-κB, and 5′-GAAAACTGAAAGGGAGAACTGAAA-3′ for IRF-3. The oligonucleotides were biotinylated at the 5′ end. Crude nuclear protein samples (10 μg) were incubated for 20 min at room temperature in a binding reaction system containing 1.5 μL 10× binding buffer, 1.5 μL poly(dI-dC) (1.0 μg/μL), and ddH2O, to a final volume of 14.4 μL. Next, 0.6 μL (300 fmol) of the probe was added, and the reaction was incubated for 20 min at room temperature. Where indicated, 2 μL of specific competitor cold oligonucleotides in 100× competing buffer was added before the labeled probe, and the reaction was incubated for 20 min. Protein-DNA complexes were resolved by electrophoresis at 4°C on a 6.5% acrylamide gel and subjected to autoradiography. Electrophoresis was performed on a 6.5% non-denaturing polyacrylamide gel at 175 V in 0.25×TBE (1×TBE, 89 mM Tris-HCl, 89 mM boric acid, and 5 mM EDTA, pH 8.0) at 4°C for 1 h. The gels were transferred to the bonding membrane at 394 mA in 0.5×TBE at room temperature for 40 min. After cross-linking for 10 min with a UV cross-linking apparatus (immobilization), the membrane was blocked, streptavidin-HRP labeled, washed again, and equilibrated; images were captured with an Imager apparatus (Alpha Innotech, San Leandro, CA).
Statistical analysis
The results are expressed as the mean±standard deviation (SD). The data were analyzed with Student's t-test (two-tailed). A p value of <0.05 was considered statistically significant.
Results
Persistent and potent silencing of TLR4 mRNA
To examine the silencing effect of the siRNA in EVC-304 TLR4– cells, the cells were collected, and the TLR4 mRNA was detected by RT-PCR; cells without siRNA were used as a normal control. In contrast to EVC-304 cells, the expression of TLR4 mRNA was strongly inhibited in EVC-304 TLR4– cells (Fig. 1). Similar results were obtained in three different RT-PCR reactions. Therefore, TLR4 mRNA was strongly and persistently silenced by the siRNA in the EVC-304 TLR4– cells, and these cells were used in the experiments described below.
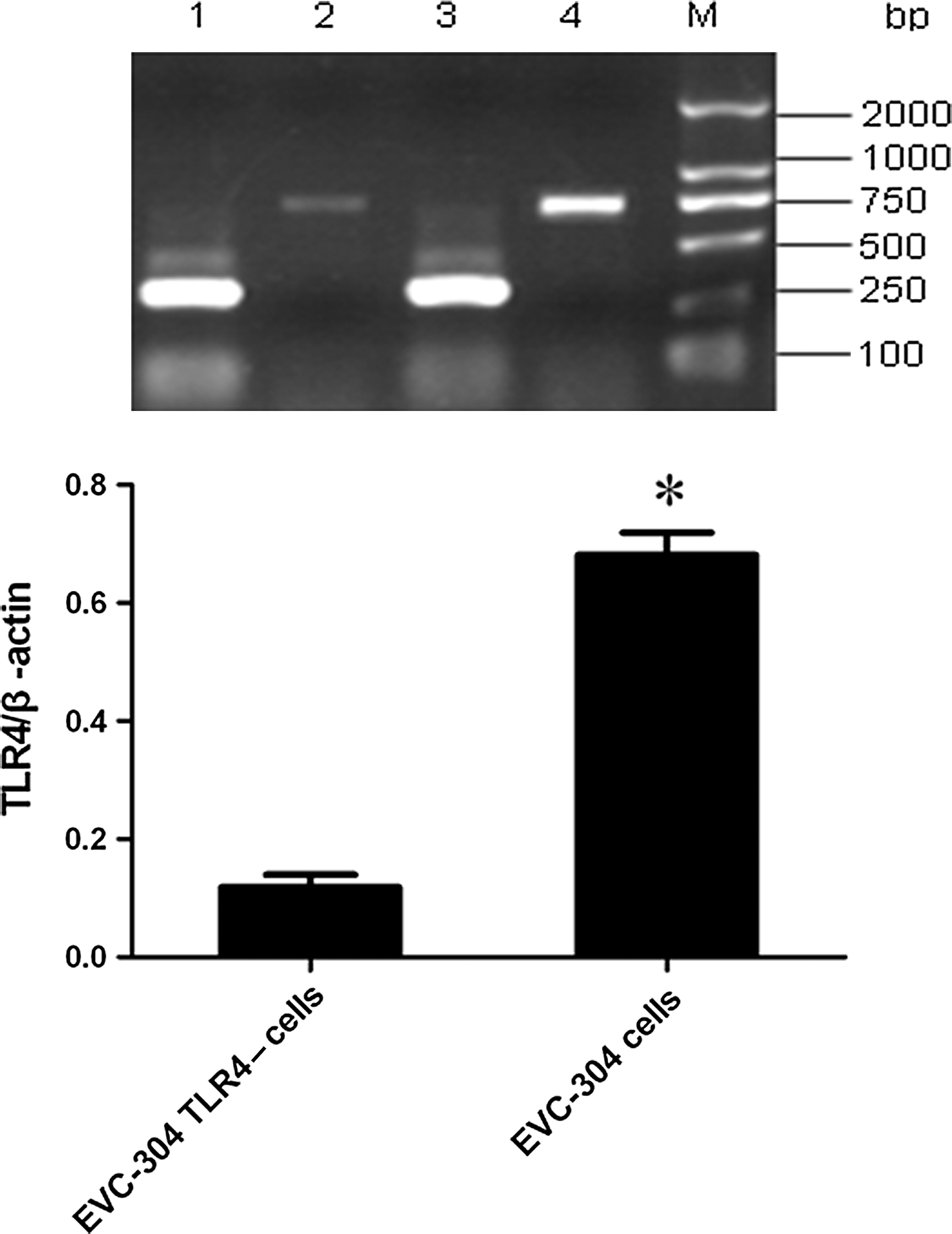
Top panel: The silencing effect of siRNA on TLR4 in EVC-304 cells (lanes 1 and 3, the expression of β-actin in both cell lines; lane 2, TLR4 mRNA from EVC-304 TLR4– cells; lane 4, TLR4 mRNA from EVC-304 cells; lane M, DNA marker). Bottom panel: The data are presented as the mean plus standard deviation from three independent experiments (*p<0.001 compared with EVC-304 TLR4-cells).
HTNV infection upregulates TRIF in EVC-304 cells
To determine the signaling pathway of TLR4 in HTNV-infected EVC-304 cells, quantitative real-time PCR and Western blotting were used to detect the expression of TRIF and MyD88, which are important adaptor proteins in TLR4 signaling. Quantitative real-time PCR demonstrated that TRIF mRNA expression was significantly higher (approximately four times higher) in EVC-304 cells than in EVC-304 TLR4– cells (Fig. 2B). No obvious difference in MyD88 mRNA expression was observed between the two cell lines (Fig. 2A). Western blot assays produced similar results (Fig. 2B and D).
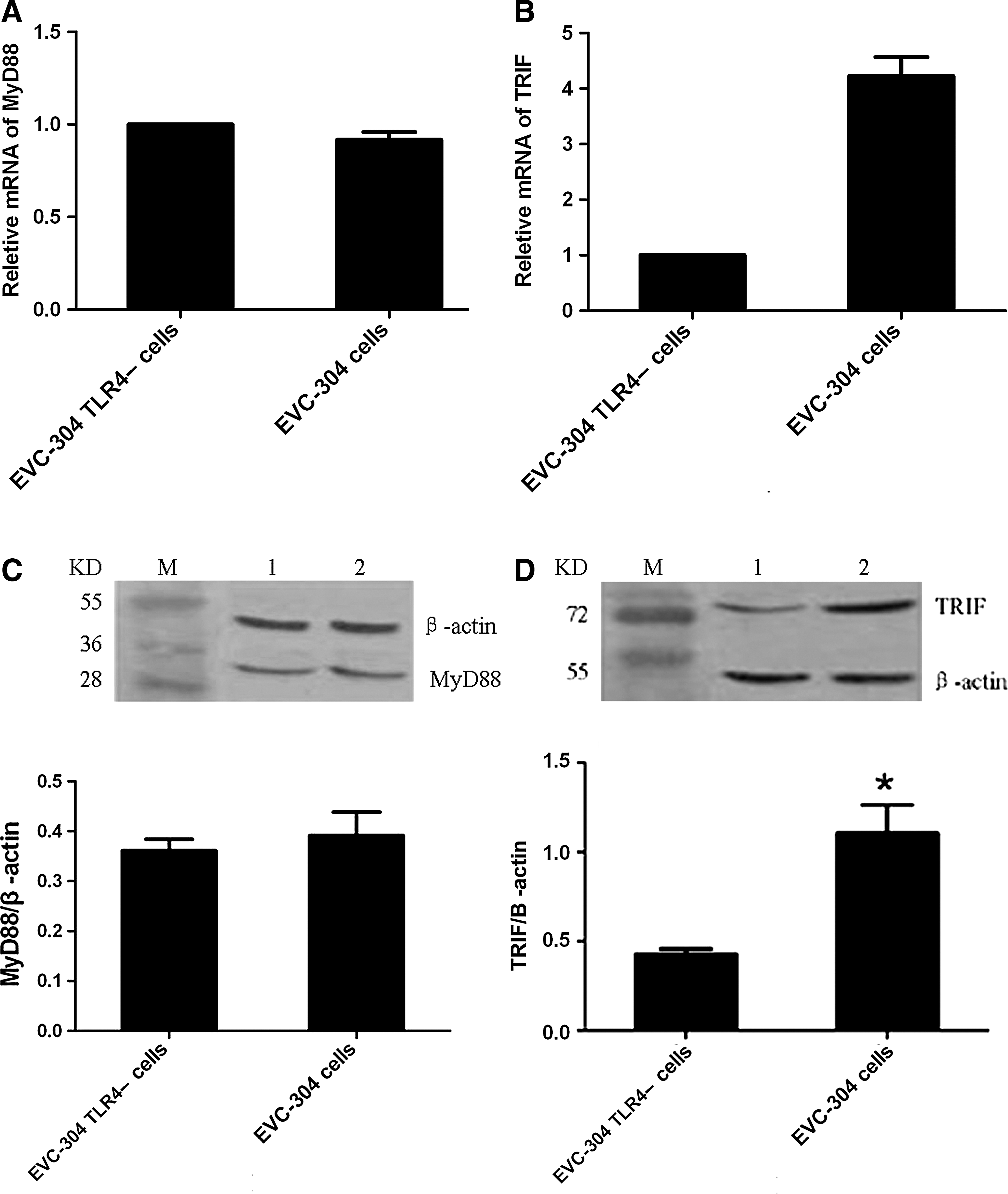
Detection of MyD88 and TRIF in HTNV-infected EVC-304 cells and EVC-304 TLR4– cells by real time PCR (
TLR4 mediates the nuclear translocation of NF-κB and IRF-3
HTNV was used to infect EVC-304 and EVC-304 TLR4– cells. Complete RPMI 1640 medium was used as a mock-infected control, and cells stimulated with LPS were used as a positive control. The nuclear translocation of NF-κB (Figs. 3A–F and 4A), and IRF-3 (Figs. 3G–L and 4B) was determined at different time points (1, 3, and 6 h) by IFA and EMSA.
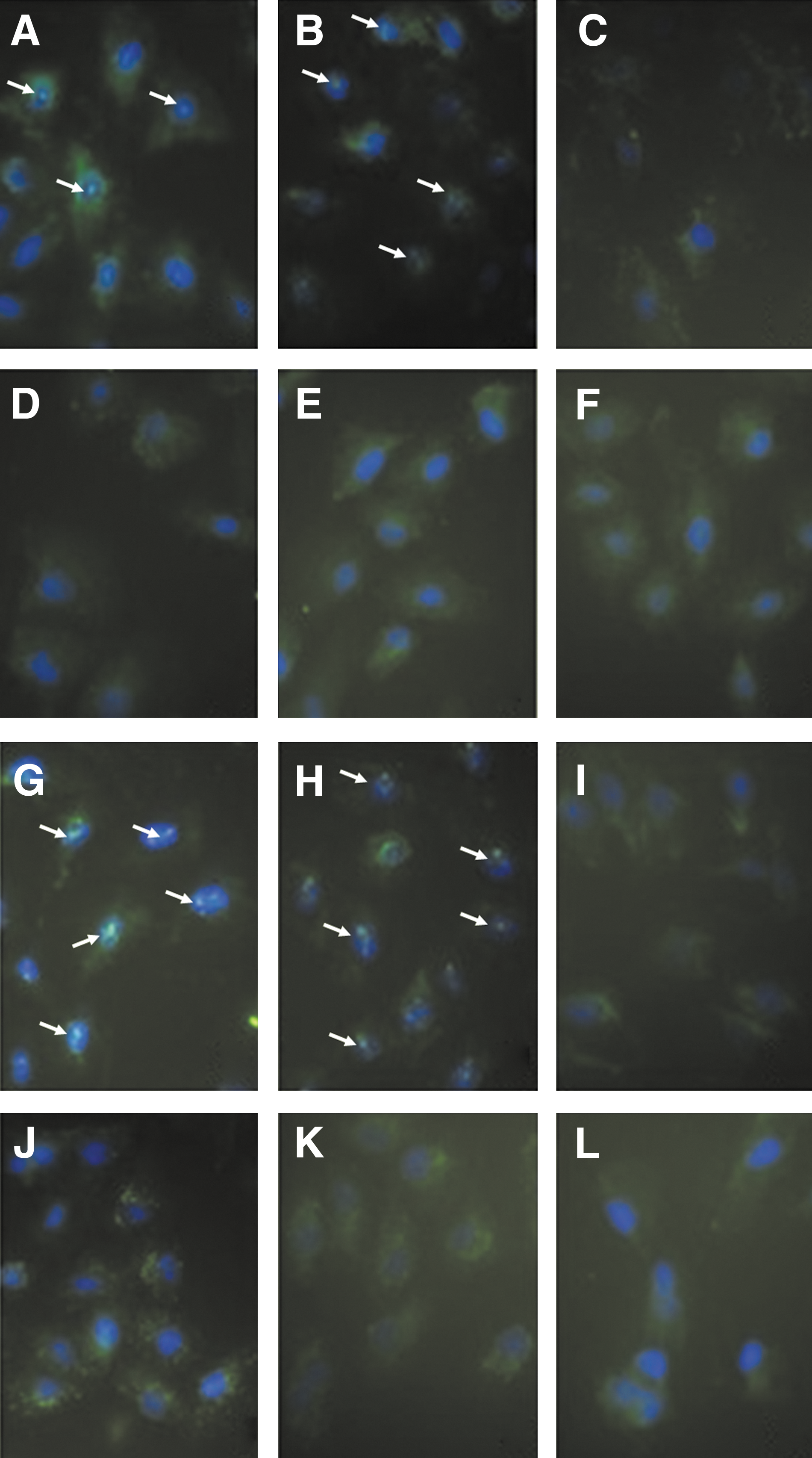
Nuclear translocation of the transcription factors NF-κB and IRF-3. (
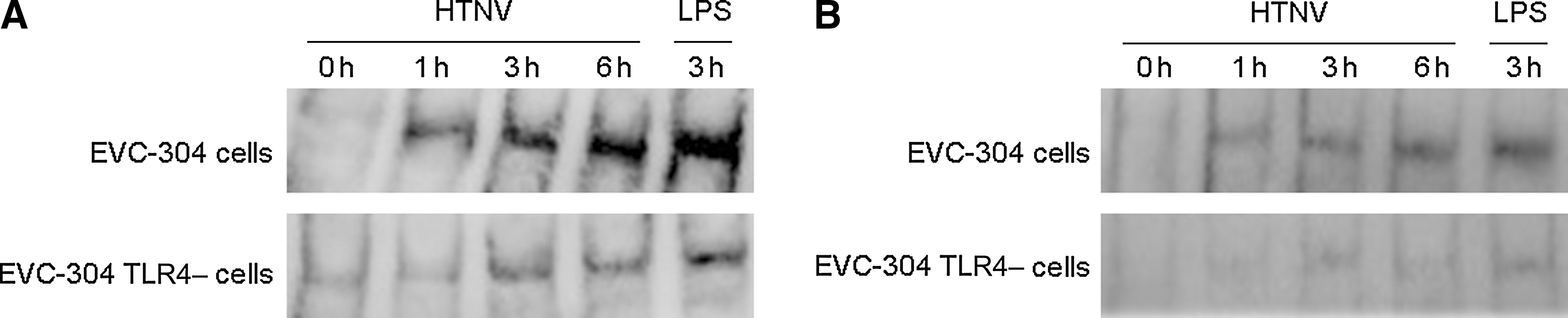
Detection of the nuclear translocation of the transcription factors NF-κB and IRF-3 by EMSA. Following an overnight incubation, EVC-304 cells and EVC-304 TLR4– cells were infected with HTNV 76–118. At different time points (0, 1, 3, and 6 h), the nuclear translocation of NF-κB (
Both NF-κB and IRF-3 were detected by EMSA in the nuclear protein extracts of HTNV-infected EVC-304 cells at these time points, and translocation into the nucleus increased with time up to 6 h (Fig. 4). Less NF-κB and IRF-3 were detected in the nuclear proteins from HTNV-infected EVC-304 TLR4– cells compared with HTNV-infected EVC-304 cells (Fig. 4). These results were verified with IFA, which yielded similar results; because the clearest data were obtained at 6 h (Fig. 3), the 0-, 1-, and 3-h data are not shown. The EMSA and IFA data indicate that TLR4 may mediate the nuclear translocation of NF-κB and IRF-3 in HTNV-infected EVC-304 cells.
Discussion
Innate immunity acts as a sentinel against microbial pathogen invasion and is activated immediately following the recognition of diverse PAMPs by various pattern-recognition receptors (PRRs). TLRs, which are important PRRs, induce effective immune responses and trigger the release of inflammatory cytokines and type I interferons for host defense (2,4,17,24). 12 TLRs have been identified in mammals (2,4,17,24), and they have been implicated in the recognition of a diverse range of microbial pathogens, including bacteria, fungi, viruses, and protozoa (3).
In a previous study, five TLRs (TLR2, TLR3, TLR4, TLR7, and TLR9) were detected in HTNV-infected vascular endothelial cells; however, only the expression of TLR4 was upregulated (10). Although increased capillary permeability is the main pathological physiological symptom of HFRS, hantavirus is not directly cytopathic to infected endothelial cells (15,29,35,42). Therefore, it is more likely that HFRS is caused by cytokines/chemokines than by endothelial cell injury. TNF-α, IL-6, and IFN-β are three important cytokines that contribute to the increased permeability of endothelial cells (21,26,31,43). This study shows that in HTNV-infected vascular endothelial cells, TLR4 may mediate the increased expression of TNF-α, IL-6, and IFN-β (10), cytokines that are closely associated with HFRS. Therefore, TLR4, as well as the expression of relevant downstream genes, may play important roles in the different stages of HFRS.
TLRs are trimodular in structure; the intracellular C-terminal domain of TLRs is known as the TIR domain, and exhibits homology with the C-terminal domain of the IL-1 receptor. When activated, this domain is required for the interaction and recruitment of various adaptor molecules to initiate the downstream signaling pathway (17). There are five TIR adaptor proteins: MyD88, TIRAP, TRIF, TRAM, and SARM, and different TLRs use different combinations of adaptor proteins to initiate downstream signaling. Interestingly, TLR4 is the only TLR that utilizes all five of these adaptor proteins (23,27). Following stimulation with LPS, TLR4 transfers the activated signal via two pathways: a MyD88-dependent pathway and a MyD88-independent (TRIF-dependent) pathway. MyD88, which recruits other death domain-containing molecules through homotypic interactions, is an important TIR-containing adaptor protein that mediates MyD88-dependent signaling. This adaptor activates the transcription factors NF-κB, AP-1, and IRF-5, and is responsible for the expression of proinflammatory cytokines. TRIF, an important adaptor protein in the MyD88-independent pathway, signals the induction of type Ι interferons by activating the transcription factor IRF-3, as well as NF-κB and AP-1 (23). Therefore, we investigated the role of MyD88 and TRIF in TLR4 signaling in HTNV-infected vascular endothelial cells. In this study, the expression of TRIF was upregulated in HTNV-infected EVC-304 cells compared with HTNV-infected EVC-304 TLR4– cells. However, we consistently observed that there was no significant difference in the expression of MyD88 in these two cell lines. In contrast to LPS stimulation, a MyD88-independent TLR4 signaling pathway may play a role in HTNV infection.
Under normal conditions, the transcription factors NF-κB and IRF-3 are primarily sequestered and inactivated in the cytoplasm (6). Upon activation, these molecules enter the nucleus and activate target gene expression by inducing the assembly of active promoters and recruiting coactivators to target gene promoters (7,34). Depending on the duration of the stimulus, NF-κB and IRF-3 undergo repeated rounds of nuclear entry and cytoplasmic redistribution in a dampened oscillatory manner (6). In HTNV-infected EVC-304 cells, we observed the nuclear translocation of NF-κB and IRF-3 at 1, 3, and 6 h after infection with HTNV, but a similar phenomenon was not observed in TLR4– cells. Previous studies have shown that TRIF plays a key role in the activation of IRF-3, and in the late-phase activation of NF-κB (23). Therefore, HTNV may induce the TLR4-mediated activation of NF-κB and IRF-3 by a MyD88-independent signaling pathway. In LPS-stimulated cells, both MyD88 and TRIF are recruited and activate TLR4 signaling. It is possible that LPS and HTNV activate TLR4 in different ways. In MyD88 KO macrophages, NF-κB was detected in the nuclear extract, but the cells did not secrete detectable levels of IL-6 and TNF-α in response to LPS (13), in contrast to our findings. Therefore, it is possible that MyD88 and TRIF play different roles in response to LPS, but TRIF may play a central role in the response to HTNV. Moreover, MyD88 may mediate functions in HTNV-infected cells, the details of which are unknown.
Conclusion
Since HTNV was first isolated in 1978 (19), it has been the subject of intensive research; however, the pathogenesis of HFRS is unknown. Increasing evidence points to an inflammatory response as the main source of the pathological changes associated with HFRS. Our study suggests that TLR4 may play an important role in HTNV pathogenesis, and functions through a MyD88-independent pathway. Although more work is required to fully understand the mechanisms of this infection, the data presented in this study provide the basis for future studies.
Footnotes
Author Disclosure Statement
No conflicting financial interests exist.
Acknowledgments
We thank Dr. Guangyu Li from the University of Texas Medical Branch for carefully reading the manuscript and improving the English writing. This work was supported by the National Basic Research Program of China (973 Program, no. 2012CB518905), and National Natural Science Foundation of China (no. 30671967 and 81071370).