
Abstract
Epstein-Barr virus (EBV), like many other persistent herpes viruses, has acquired numerous mechanisms for subverting or evading immune surveillance. This study investigates the role of secreted EBV-encoded BARF1 protein (sBARF1) in creating an immune evasive microenvironment. Wild-type consensus BARF1 was expressed in the human 293 cell line and purified. This native hexameric sBARF1 had inhibitory capacity on macrophage colony stimulating factor (M-CSF)-stimulated, and not on granulocyte macrophage-colony stimulating factor (GM-CSF)-stimulated growth and differentiation of myeloid cells. Antibodies specific to hexameric sBARF1 were able to block this effect. M-CSF was shown to interact with sBARF1 via the protruding N-terminal loops involving Val38 and Ala84. Each BARF1 hexamer was capable of binding three M-CSF dimers. Mutations in the BARF1 loops greatly affected M-CSF interaction, and showed loss of growth inhibition. Analysis of the activation state of the M-CSF receptor c-fms and its downstream kinase pathways showed that sBARF1 prevented M-CSF-induced downstream phosphorylation. Since M-CSF is an important factor in macrophage differentiation, the effect of sBARF1 on the function of monocyte-derived macrophages was evaluated. sBARF1 affected overall survival and morphology and significantly reduced expression of macrophage differentiation surface markers such as CD14, CD11b, CD16, and CD169. Macrophages differentiating in the presence of sBARF1 showed impaired responses to lipopolysaccharide and decreased oxygen radical formation as well as reduced phagocytosis of apoptotic cells. In conclusion, EBV sBARF1 protein is a potent decoy receptor for M-CSF, hampering the function and differentiation of macrophages. These results suggest that sBARF1 contributes to the modulation of immune responses in the microenvironment of EBV-positive carcinomas.
Introduction
Computer-assisted amino acid sequence analysis of sBARF1 indicated weak homology with a conserved region found in several members of the tyrosine kinase family, including the receptor for macrophage colony stimulating factor (M-CSF), platelet-derived growth factor (PDGF) receptor, fibroblast growth factor (FGF) receptor, and hepatocyte growth factor (HGF) receptor (6,52). The homology with these receptors is restricted to a conserved structural region not involved in the ligand binding domains (52). Full-length BARF1 has homology with the T-cell receptor co-stimulatory molecule CD80 (54). Prior studies with monomeric Fc-tagged BARF1 indicated that BARF1 is a viral scavenger of M-CSF, binding all three isoforms, and acting as a functional antagonist for M-CSF (36,52). However, subsequent studies on the structure of secreted BARF1 revealed a hexameric molecule, which may differ in structure and function from monomeric Fc-tagged BARF1 (18,54,56).
M-CSF is synthesized by many cell types, including endothelial cells, fibroblasts, bone marrow stromal cells, osteoblasts, and keratinocytes. Serum levels of M-CSF in normal adults range from 1.7–8.4 ng/mL (22), but are elevated during pregnancy, infection, lymphomas, and breast, ovary, and endometrial carcinomas (5,7,22,49). High-level secreted BARF1 was observed in serum samples of NPC patients (19). M-CSF is a pleiotropic growth factor with many roles in the immune system; M-CSF is important for regulating the viability, proliferation, and differentiation of mononuclear phagocytes from monocytes to macrophages, but also for the differentiation of Langerhans and other dendritic cell (DC) subsets (5,20,28,35,40,48). The most appreciated function of M-CSF relates to the induction of monocyte differentiation into resident macrophages (M2 macrophages) (14,40).
EBV-infected monocytes have been suggested as a vehicle for virus transmission between the blood compartment and oral epithelium, and EBV infection in vitro alters monocyte function by affecting innate inflammatory functions (13,30,47,55). Following differentiation into DCs, monocytes become resistant to apoptosis caused by EBV infection (13). M-CSF inhibition by secreted BARF1 might be instrumental for EBV in restricting maturation of macrophage effector functions, thus ensuring that these immune cells support dissemination of viral progeny rather than elimination of the internalized or bound virus. In NPC, lymphocyte infiltration does not affect prognosis (23,26), but infiltration of NPC with mononuclear cells is correlated with a higher survival rate (12,39), indicating their involvement in tumor cell eradication. An EBV-infected tumor cell, expressing viral antigens that can be recognized, might thus benefit from local myeloid suppression resulting from M-CSF inhibition by BARF1. Humoral immune responses to BARF1 can be detected, but are relatively low (18). BARF1 was recently shown to trigger both CD4 and CD8 cellular immune responses in NPC patients, providing a basis for specific immunotherapy (29). However, the lymphocytes from the NPC tumor environment are functionally impaired (23,26). By binding and functionally inhibiting M-CSF, sBARF1 might interfere with myeloid sentinel functions, and thus contribute to an immunological silence facilitating immune escape. Clearly, more insight into how EBV manipulates the immune system is necessary for the progress of advanced immune therapy.
In this study, an NPC-sequence-derived native hexameric sBARF1 protein was used to assess its biological effects as a decoy receptor for M-CSF, and to evaluate its effects on cells of myeloid lineage. We showed that M-CSF-induced macrophage differentiation, function, and survival is severely impaired in the presence of native sBARF1, further characterizing it as a viral immune suppressive factor with possible effects on both viral dissemination and cancer immune escape. Mutations in the N-terminal loops of sBARF1 demonstrated the specificity of the sBARF1/M-CSF interaction, indicating that future therapies should be aimed at this site.
Materials and Methods
Cell culture
Human cytokine-dependent MUTZ-3 cells were obtained from the German collection of microorganisms and cell cultures DSMZ (Braunschweig, Germany), and cultured in Costar 12-well tissue culture plates (Corning, Amsterdam, Netherlands), at 0.4 million cells in 2 mL MEM-α containing 10% FCS, 100 U/mL sodium penicillin, 100 μg/mL streptomycin sulfate, 2 mM L-glutamine (P/S/G), and 50 μM β-mercaptoethanol, supplemented with 10% conditioned medium from the human renal carcinoma cell line 5637 (31,43,44). Peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats of healthy donors by Ficoll density gradient centrifugation. CD14+ monocytes were isolated from PBMCs by magnetic sorting with anti-CD14 microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany), according to the manufacturer's protocol. Monocytes were differentiated into monocyte-derived macrophages (MoΦ) in 5–7 days in IMDM medium (Invitrogen, Carlsbad, CA) supplemented with 10% FCS, P/S/G, and 10 ng/mL human M-CSF (Miltenyi Biotec). Where indicated, M-CSF was preincubated with purified native human sBARF1 (200 ng/mL unless indicated otherwise) (18) for 1 h at room temperature before adding it to the monocytes to create sBARF1-treated macrophages (sBARF1-treated MoΦ).
sBARF1 production
Soluble hexameric BARF1 protein (sBARF1), cloned from a NPC-derived EBV isolate and expressed in HEK293 human epithelial cells, was purified to homogeneity from serum-free supernatants by lectin-affinity chromatography as previously described (18,21).
Site-directed mutagenesis
M-CSF binding site mutants of BARF1 were created using the Quickchange Lightning multi-site-directed mutagenesis kit according to the manufacturer's instructions (Stratagene, Santa Clara, CA), to incorporate specific mutations into the original pcDNA4-BARF1 expression vector. Mutant sBARF1 was produced and purified exactly the same as the wild-type sBARF1.
Immunoprecipitation
Rabbit polyclonal antibodies specifically reactive with native hexameric BARF1 were covalently bound to Protein-G beads (GE Healthcare, Waukesha, WI) using 6 mg/mL dimethylpimilimidate in 0.2 M Na2B4O7•10H2O at pH 9 for 1 h at room temperature, followed by three wash steps with 0.2 M ethanolamine. sBARF1 and attached protein was immunoprecipitated (IP) from medium by anti-BARF1 beads, eluted with 0.2 M glycine (pH 3.0), and neutralized with 3 M Tris (pH 8.0).
SDS-PAGE and Western blot
Cells were lysed in IP buffer containing Protease Inhibitor Cocktail (Roche, Basel, Switzerland) and sonicated. Cell debris was removed by centrifugation and protein concentration was determined using the BCA Protein Assay kit (Pierce, Rockford, IL). Cell extracts and IP samples were diluted in 2× loading buffer (Bio-Rad, Hercules, CA) with β-mercaptoethanol, denatured for 5 min at 95°C, and separated on a 12.5% SDS-PAGE gel. After transferring to Hybond ECL nitrocellulose membranes (GE Healthcare), the membranes were blocked in PBS with additional Tween-20 (PBST) containing 3% non-fat dried milk for 1 h at room temperature. Anti-M-CSF antibodies (1:500; Santa Cruz Biotechnology, Santa Cruz, CA), or anti-BARF1 4A6 antibodies (1:100) (18), were incubated overnight at 4°C in PBST with 5% BSA. After three wash steps with PBST, the appropriate HRP-labeled secondary antibodies (Dako, Glostrup, Denmark) were incubated 1:2000 in 3% milk for 1 h, and the antibody was removed by three wash steps with PBST, followed by visualization with ECL (GE Healthcare). Pathscan Multiplex Western Cocktail (#5301) was used to visualize phosphorylated MAPK, Akt, and phosphorylated M-CSF receptor (#3151; Cell Signaling Technology, Danvers, MA), according to the manufacturer's protocol.
Viability assay
Approximately 100,000 MoΦ were cultured in 200 μL medium per well in 96-well plates. Viability was determined with a cell proliferation kit (MTT; Roche), according to the manufacturer's protocol. Absorbance was measured with a Tecan Spectafluor at 590 nm.
Giemsa and nitroblue tetrazolium staining
Following methanol fixation, cells were stained with Giemsa solution (Merck Sharp & Dohme, Whitehouse Station, NJ) 1:5 in H2O for 1 h. To determine the production of free radicals upon lipopolysaccharide (LPS) stimulation, MoΦ were pre-sensitized with 200 U/mL human interferon-γ (Miltenyi Biotec) for 2 h. Subsequently 1 μg/mL LPS (E. coli strain 055:B5; Dako) and 0.04% nitroblue tetrazolium (NBT; Sigma-Aldrich, Buchs, Switzerland) were added and incubated for 4 h. After treatment, the cells were fixed and light microphotographs (40×) were taken with Leica software (Wetzlar, Germany).
Flow cytometry
MoΦ were harvested with 0.05 mM EDTA (Merck Sharp & Dohme) in PBS and washed with fluorescence-activated cell sorter (FACS) buffer (0.2% BSA and 0.002% NaN3 in PBS), before staining with monoclonal antibodies specific for macrophage surface markers for at least 30 min at 4°C. The monoclonal antibodies used were fluorescein isothiocyanate (FITC)-conjugated IgG1 (isotype control), phycoerythrin (PE)-conjugated against IgG1 (isotype control), FITC-conjugated against CD14, CD16, and CD64, PE-conjugated against HLA-DR, CD11b, CD11c, CD36, and CD1a (BD Biosciences, Franklin Lakes, NJ), and FITC-conjugated against CD169 (Abcam, Cambridge, U.K.). After washing, the cells were analyzed on a FACSCalibur II system (BD Biosciences). The results were analyzed with CellQuest Pro software.
Phagocytosis assay
MoΦ were cultured on glass chamber slides during differentiation. Jurkat cells were dyed fluorescent green using PKH67 dye (Invitrogen) according to the manufacturer's protocol, 1 day before lethal irradiation with 50 Gy. After 5 h, apoptotic cells were added to the macrophages. After 1.5 h incubation, the macrophages were washed four times with ice-cold PBS and fixed with 4% paraformaldehyde at 4°C for 30 min. To visualize macrophage contours, the cells were washed three times with PBS, permeabilized with 0.1% Triton-X100 in PBS, and incubated with 1:40 rhodamine phalloidin (Sigma Aldrich) in PBS and 5% BSA for 1 h at room temperature. After washing, the slides were cover-slipped, and pictures were taken using a Leica confocal microscope.
Data analysis
The Wilcoxon signed rank test was performed using GraphPath-4 statistical software (La Jolla, CA), and p values<0.05 were considered significant.
Results
Secreted BARF1 protein binds and functionally inhibits M-CSF
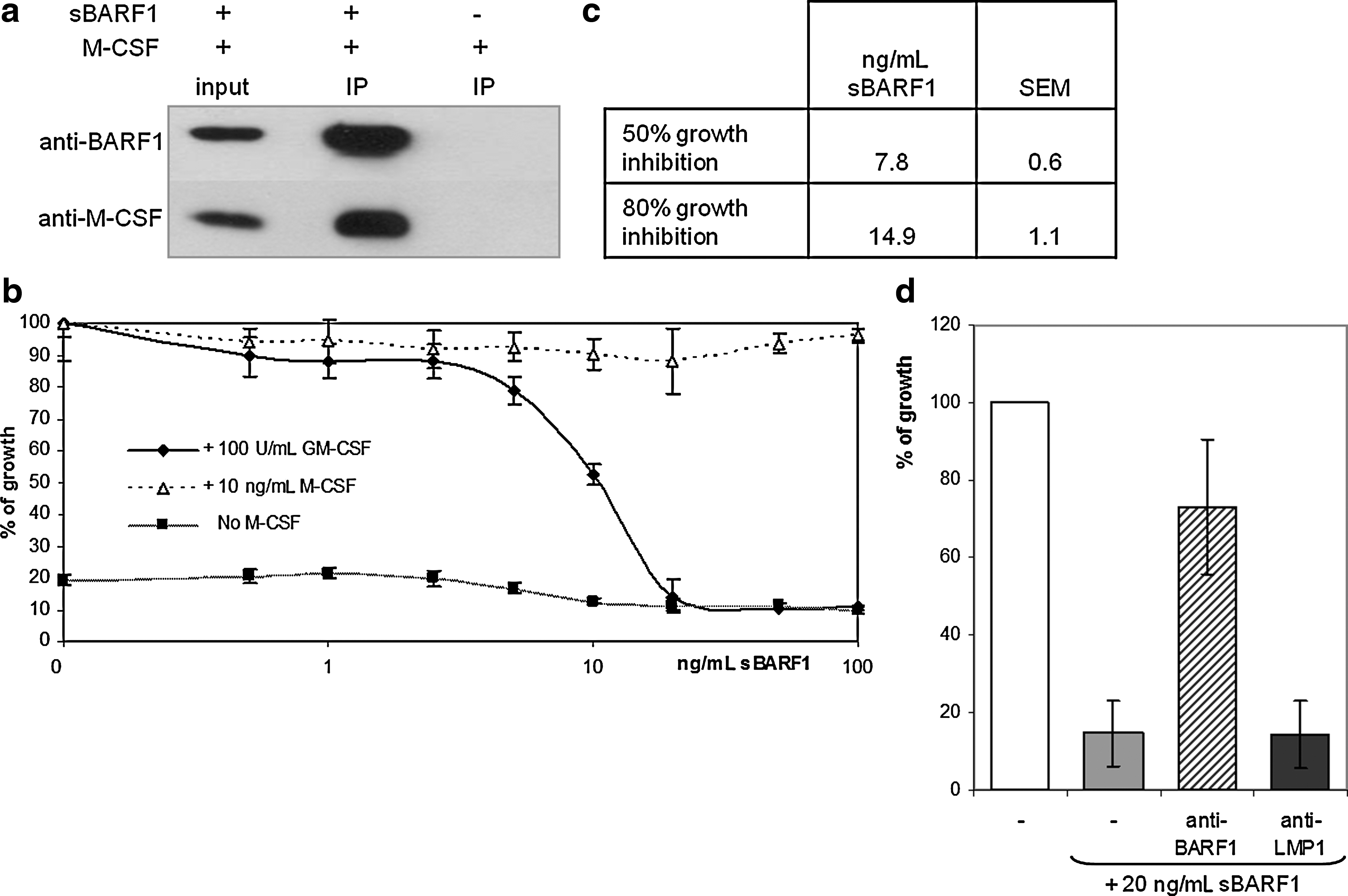
To evaluate whether the native secreted hexameric form of BARF1 could bind M-CSF, co-immunoprecipitations were performed in DMEM supplemented with purified sBARF1 or M-CSF or both. Protein-G beads covalently linked to anti-sBARF1 antibody, recognizing the hexameric form of sBARF1, were used to precipitate BARF1 from medium with or without M-CSF. Immunoblot analysis revealed that M-CSF was specifically precipitated when BARF1 was present in the medium (Fig. 1a), indicating a direct binding of sBARF1 to M-CSF.
(
The human myeloid cell line MUTZ-3 was used to evaluate whether sBARF1 binding to M-CSF functionally interfered with cell proliferation. These cells are dependent on human cytokines for in vitro growth, and thus can be used to evaluate functional cytokine levels. MUTZ-3 cells were cultured in the presence of 10 ng/mL M-CSF, which was pre-incubated with sBARF1 at different concentrations. As shown in Fig. 1b, sBARF1 was able to block M-CSF-dependent growth of MUTZ-3 cells in a dose-dependent manner. Concentrations higher than 17 ng/mL of purified sBARF1 were able to block 10 ng/mL M-CSF, equaling one sBARF1 hexamer binding three M-CSF dimers. In addition, MUTZ-3 cells can also be maintained in the presence of granulocyte macrophage-colony stimulating factor (GM-CSF), which has a tertiary structure similar to M-CSF, but a different quaternary structure and receptor interaction. Of note, sBARF1 was not able to block GM-CSF-dependent cell proliferation, demonstrating its binding specificity for M-CSF, and confirming that sBARF1 by itself has no toxic or harmful effect (Fig. 1b). The proliferation of MUTZ-3 cells in the presence of sBARF1 could be restored by pre-incubation with a neutralizing antibody against BARF1 (Fig. 1d). These results showed that sBARF1 is able to functionally inhibit M-CSF, but not GM-CSF, and that this can be neutralized by specific antibodies.
Structural basis of the interaction between M-CSF and sBARF1
Sequence analysis revealed structural homology of sBARF1 with several growth factor receptors, but gave no indication which parts of the sBARF1 hexamer are responsible for M-CSF binding. M-CSF is a disulfide-linked dimer with each monomer consisting of a four-alpha-helix bundle and an anti-parallel beta sheet (38). The M-CSF receptor binds the M-CSF dimer at its alpha-helix flat face, but the affinity is weak. Upon binding to the receptor, the M-CSF dimer undergoes conformational changes, resulting in dimerization and activation of the receptor (4). Crystallization results of the sBARF1/M-CSF complex (unpublished data) indicated that in contrast to the M-CSF receptor, the BARF1 hexamer binds the M-CSF dimer at the beta sheets, with Val38 and Ala84, located in two protruding N-terminal loops, as the main interacting residues on sBARF1. To confirm these findings, Val38Glu and Ala84Glu single mutants, a double mutant, and a mutant in which the loop residues 37–40 were mutated to glycine, were generated. The double mutant and the loop mutant were secreted as hexamers in the culture medium of transfected cells (Fig. 2a). The intracellular loop mutant resulted in relatively less glycosylated BARF1, possibly relating to folding difficulties during intracellular trafficking of the translated product. The single mutants were able to bind M-CSF, but both the double and the loop mutant were disabled in M-CSF binding (Fig. 2b), and showed reduced inhibition of M-CSF-dependent growth of MUTZ-3 cells compared to wild-type (WT) BARF1 (Fig. 2c).

Site-directed mutants of sBARF1 reveal the M-CSF binding site. (
M-CSF receptor downstream signaling is inhibited by sBARF1
The effect of sBARF1 in the M-CSF receptor downstream signaling pathway, involving tyrosine kinases PI3-K and STAT (14,15,34), was evaluated. Five minutes after exposing serum-starved MUTZ-3 cells to M-CSF, phosphorylated (active) M-CSF receptor, Akt, and MAPK were detected (Fig. 3a). Pre-incubating M-CSF with sBARF1 prevented this activation of the M-CSF-receptor signaling pathway, whereas pre-incubation of sBARF1 with a neutralizing antibody restored the signaling through the M-CSF receptor (Fig. 3a). Since macrophages differentiate in the presence of M-CSF and express the M-CSF receptor, downstream signaling was also evaluated in serum-starved MoΦ. Short-term M-CSF exposure increased MAPK phosphorylation, whereas pre-incubation of M-CSF with sBARF1 reduced this MAPK phosphorylation (Fig. 3b). Thus kinase pathways, activated by M-CSF binding to its receptor, were negatively modulated when M-CSF was scavenged by sBARF1.
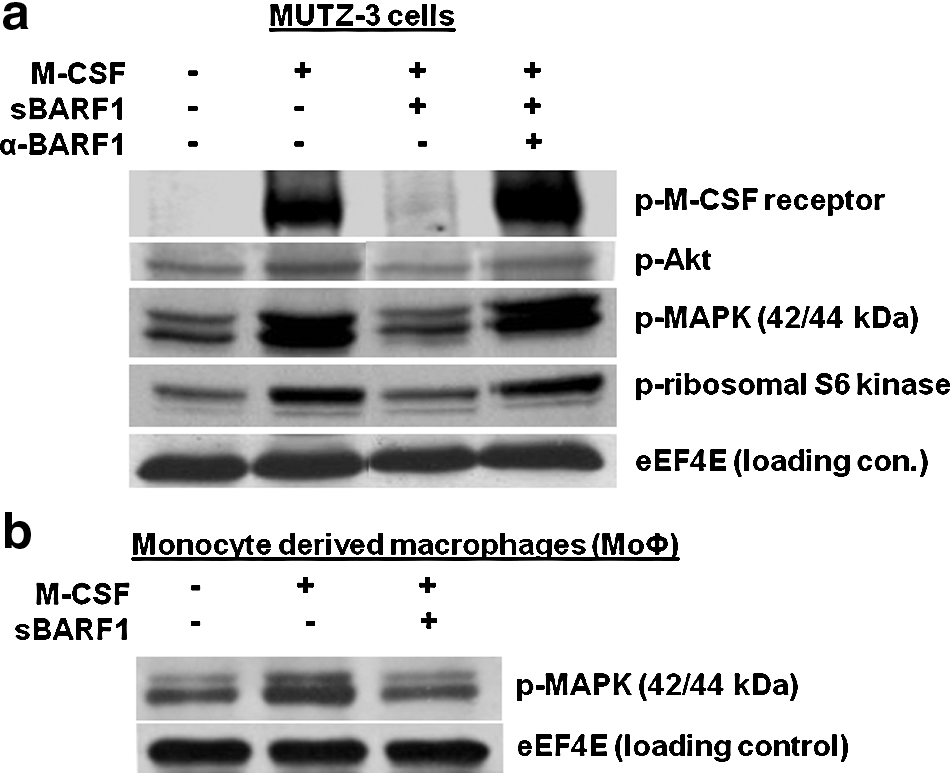
M-CSF receptor downstream signaling is inhibited by sBARF1. (
Native sBARF1 negatively influences cell morphology and viability during macrophage differentiation
M-CSF receptor (c-fms) is expressed on both macrophages and DCs (45,46) and their precursors (11,37). The importance of M-CSF in DC differentiation is reflected by M-CSF-deficient mice, which have a two- to threefold reduction in splenic DCs (28). Since M-CSF is essential for macrophage differentiation (14,40), the effect of sBARF1 on this process was evaluated using CD14+ monocytes from healthy donors. Differentiated MoΦ were characterized by enlarged cytoplasm and irregular plasma membranes with pseudopodia. After 5 d of differentiation in the presence of M-CSF, MoΦ showed the typical morphology of differentiated macrophages. In contrast, sBARF1-treated MoΦ remained small in size with a rounded morphology. Moreover, the addition of sBARF1 resulted in lower viability in the cultures, as indicated by non-adherent dead cells that were washed away by the Giemsa staining (Fig. 4a).

Native sBARF1 negatively influences macrophage morphology and viability during differentiation. Monocytes were differentiated for 6 d with 10 ng/mL M-CSF with or without 200 ng/mL sBARF1. (
To substantiate the observation that sBARF1 negatively influenced macrophage differentiation and induced cell death, a viability test was performed, with a read-out based on mitochondrial activity (i.e., MTT test). In this regard it is important to note that MoΦ acquire higher mitochondrial activity during differentiation (33). The definition of viability in these assays is both an increase in cell numbers and in the level of differentiation. When viability of M-CSF-treated MoΦ cultured in the absence of sBARF1 measured at day 6 was set at 100%, cells treated with sBARF1 showed a significant reduction in viability compared to normal differentiated cells (p=0.03; Fig. 4b). This effect was dose-dependent, and the sBARF1-induced reduction of viability occurred at 20 ng/mL and higher (Fig. 4c), and could be detected as early as day 3 of differentiation (Fig. 4d). Compared to the untreated control group, for which the viability increased upon differentiation, the viability of the sBARF1-treated cells decreased, suggesting cell death (Fig. 4d), which is in accord with our initial microscope observations (Fig. 4a).
Effects of sBARF1 on macrophage surface marker expression
To further evaluate the effect of sBARF1 treatment on macrophage differentiation, sBARF1-treated and sBARF1-untreated MoΦ were harvested on days 5–7 with EDTA and stained for key macrophage markers (i.e., CD11b, CD11c, CD14, CD16, CD36, CD64, CD169, and HLA-DR). Only adherent cells were harvested, and only viable MoΦ were selected for flow cytometric analysis. sBARF1-cultured MoΦ showed a significantly reduced level of expression of CD14, a pattern recognition co-receptor for bacterial lipopolysaccharide, and CD11b, a integrin subunit of macrophage-antigen 1 involved in adherence. Purified sBARF1 also suppressed the expression of the FcγR CD16 and CD169, a macrophage differentiation-specific marker (Fig. 4e), HLA-DR, CD11c, CD64, and CD36, were unaffected (data not shown). These findings indicate that sBARF1 influences the differentiation of macrophages.
Impairment of oxygen radical production by sBARF1-treated macrophages
MoΦ are responsible for several different effector functions of the immune system against pathogens, which can be triggered by the TLR4 ligand LPS. Upon activation, MoΦ respond by the generation of free oxygen radicals to kill bacteria that are engulfed by phagocytosis. Oxygen radicals can be visualized by NBT, which is converted into blue crystals when oxidized. MoΦ were incubated with NBT together with 1 μg/mL LPS after being pre-sensitized with IFN-γ. The sBARF1-treated MoΦ were significantly less capable of converting NBT than normally-differentiated macrophages (p=0.01; Fig. 5a).
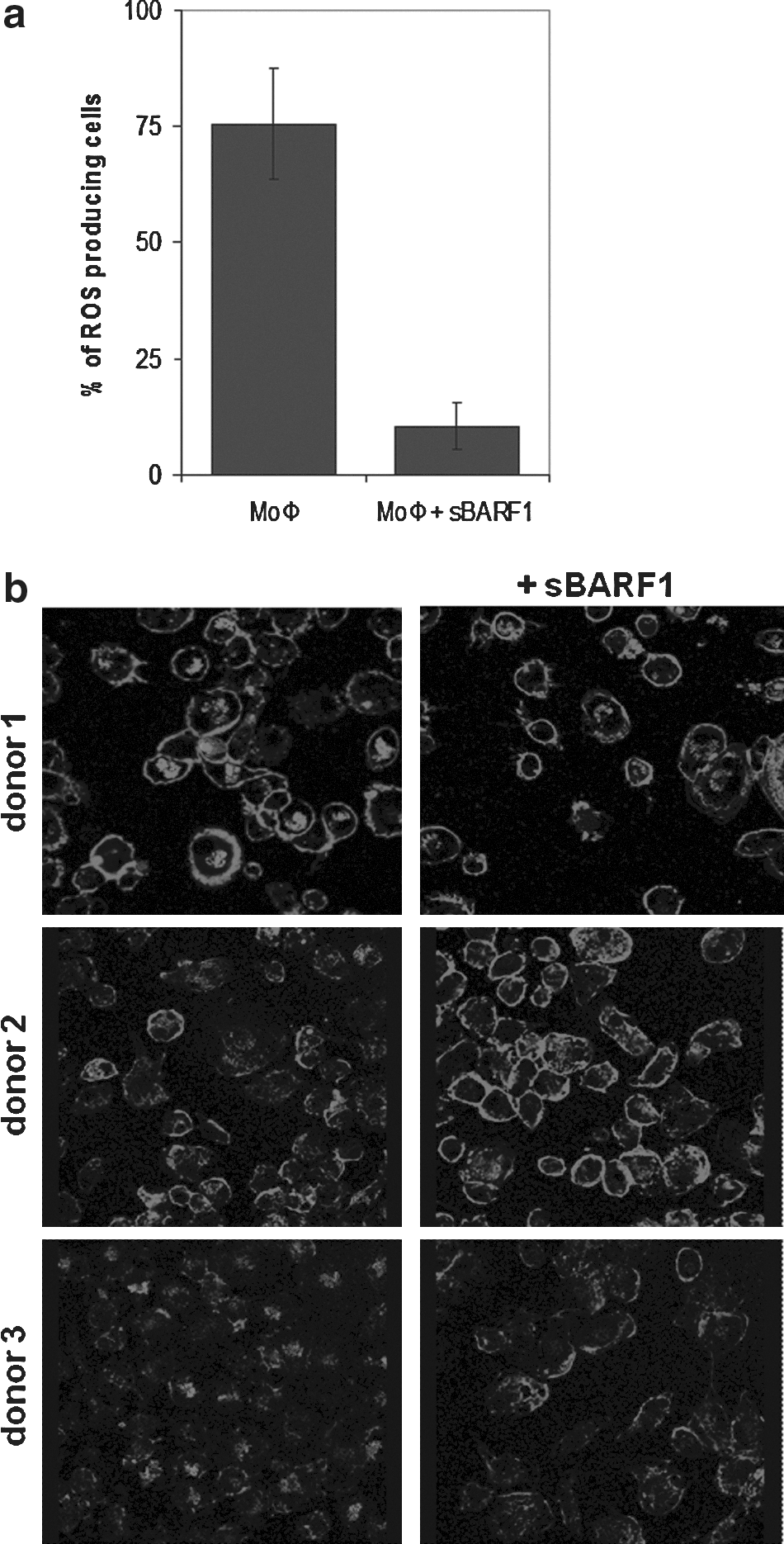
Native sBARF1 negatively influences macrophage activation and function. Monocytes were differentiated for 5 days with 10 ng/mL M-CSF with or without 200 ng/mL sBARF1. (
sBARF1 specifically affects the phagocytosis of apoptotic cells
Macrophages are known for their ability to capture and internalize particles, including pathogens, necrotic cells, apoptotic cells, and immune complexes (1). Non-specific phagocytosis was evaluated with fluorescent microbeads, and no difference between MoΦ cultured in the presence or the absence of sBARF1 was found (data not shown). To evaluate phagocytosis of apoptotic cells, PKH67-dyed Jurkat cells were lethally irradiated and added in excess to MoΦ. After incubation, the remaining Jurkat cells were washed away. Macrophages generated from three separate donors were fixed and stained with rhodamine-phalloidin to visualize actin filaments involved in phagocytosis and analyzed by confocal microscopy. In the presence of sBARF1, MoΦ phagocytosed fewer apoptotic Jurkat cells than control MoΦ (Fig. 5b), revealing a functional impairment.
Discussion
EBV, like many other persistent herpes viruses, has acquired numerous mechanisms aimed at subverting or evading immune surveillance. The secreted hexameric sBARF1 protein is produced by EBV-related carcinomas (51), and is expressed during lytic cycle replication of the virus in both epithelial and lymphoid cell types (59). Since sBARF1 has structural homology with growth factor receptors (52), it might function by scavenging cytokines as an evasion mechanism by which EBV-infected cells modulate local immune functions, especially those involving the myeloid lineage, to avoid immune-mediated eradication (6).
Prior investigations showing the inhibitory effects of BARF1 on myeloid cells were performed with Fc-tagged BARF1 (36,52). Subsequent studies showed that BARF1 in human epithelial cells is secreted as a hexameric molecule (18,54,56). The 3D-folding of the hexameric BARF1 molecule may be disturbed by the additional Fc-tag. Therefore the use of a native hexameric protein is preferred. This study showed that hexameric native sBARF1 could bind M-CSF in a direct and functionally inhibitory manner. sBARF1 binding prevented M-CSF-stimulated growth of the growth factor-dependent myeloid cell line MUTZ-3 in a linear fashion, but had no effect on GM-CSF-induced proliferation, confirming its specificity for M-CSF. Molecularly, one sBARF1 hexamer can block the activity of three M-CSF dimers, a result in agreement with the findings of Tarbouriech et al. (53). These results led us to investigate which domain at the surface of sBARF1 was responsible for M-CSF inhibition. Mutational analysis confirmed the specific interaction of sBARF1 and M-CSF, as predicted by molecular modeling based on the M-CSF/sBARF1 co-crystal structure (53). Mutants of the predicted sBARF1/M-CSF interaction sites showed that the N-terminal Val38 and Ala84 residues, located in protruding loops, are essential for M-CSF binding. sBARF1 binds M-CSF at the beta sheets, and not at the alpha helices like the M-CSF receptor (4). Since the M-CSF receptor interaction is dependent on conformational changes in the M-CSF dimer, the mechanism by which sBARF1 prevents M-CSF receptor activation might be interference with this conformational change.
M-CSF can drive mononuclear phagocyte differentiation (5,20,28,35,40), but when blocked by sBARF1, this function was inhibited. Several macrophage-related markers were negatively influenced by sBARF1, including CD14, CD11b, CD16, and CD163. In addition, sBARF1-treated MoΦ were less capable of producing free oxygen radicals upon stimulation, confirming that sBARF1-treated MoΦ were compromised in their anti-inflammatory reaction. Phagocytosis of apoptotic cells was also reduced in MoΦ treated with sBARF1. Moreover, downregulation of CD16, a Fc-receptor, is likely to negatively influence the phagocytosis of antibody-antigen complexes. All of these features are characteristic for the suppression of M2 differentiation.
Mononuclear phagocytes are exposed during their differentiation and migration to combinations of growth factors such as M-CSF, GM-CSF, IL-4, IL-34, IFN-γ, and Flt3L, each of which can contribute to their cellular phenotype (20). M-CSF is a pleiotropic growth factor with many roles in the immune system, some of which remain unknown. M-CSF is an important factor in the regulation of viability, proliferation, and differentiation of mononuclear phagocytes from monocytes to macrophages, but it also affects Langerhans cells and DCs (5,11,20,35,37,40,48). The growth factor GM-CSF skews monocytes towards M1 macrophages, and monocytes that continue to be exposed to M-CSF become M2 macrophages (40). M1 macrophages, also called inflammatory macrophages, are considered more inflammatory and are associated with type 1 cell-mediated immunity; they produce reactive oxygen species and are considered to have anti-tumor activity. M2 macrophages, also called resident macrophages, are important for phagocytosis and are the first line of antiviral defense. The inhibition of M-CSF by sBARF1 expressed during lytic replication might allow monocytes/macrophages to act as a virus transmission vehicle of internalized or bound virus, as opposed to inactivation of the virus (13,55).
Most tumor-associated macrophages (TAMs) are considered to be of the M2 type, and may even harbor tumor growth-promoting abilities. The proper differentiation of professional antigen-presenting DCs, considered to have strong potential anti-tumor activity, is also dependent on M-CSF, which has been shown in recent studies with M-CSF (receptor)-deficient mice (28).
The results presented here indicate that sBARF1 interferes with M-CSF, but not with GM-CSF action. BARF1 could thus selectively impair M2 TAMs in their development and function. This may appear counter-intuitive in terms of tumor immune escape, but TAMs do not only promote tumor growth by secreting growth factors and promoting angiogenesis (41); they can also serve as negative mediators of tumor growth by mediating anti-tumor cytotoxicity and antigen presentation (25). Since TAM density is positively correlated with the prognosis of NPC (12,39), blocking of M-CSF by sBARF1 might constitute a possible mechanism by which macrophage-mediated anti-tumor immunity is evaded.
Fc-tagged BARF1 can bind all three M-CSF isoforms, including cell surface M-CSF (52). Cell surface M-CSF on tumor cells evokes anti-tumor activity of macrophages in multiple tumor types, both innately and by linking to the adaptive arm of the immune system by antigen presentation to T cells (8,17,57). Since it is unknown whether NPC tumor cells express M-CSF, we can only speculate that BARF1 might have an effect on this level, interfering with macrophage-mediated anti-tumor immunity. Truncation of the rhBARF1 gene in a lymphocryptovirus rhesus macaque model resulted in lower efficiency of B-cell immortalization, possibly related to a loss of M-CSF inhibition (36). Future in vivo studies should be designed to explore the biological relevance of WT BARF1 in virus propagation and latency, and the effects of BARF1 modulation on myeloid cell function in the tumor area.
It is important to clarify the biological effects operating in the microenvironment of EBV-associated tumors to understand why these EBV-carrying carcinomas are not properly eliminated by the immune system. Our data reveal secreted BARF1 as a viral decoy receptor for M-CSF, functionally inhibiting M-CSF-dependent growth and activation of the M-CSF receptor pathway. Secreted BARF1 negatively influences mononuclear phagocyte differentiation, activation, and survival, which might contribute to EBV immune evasion. Therefore, the insights into the functional BARF1 M-CSF binding domains provided by this study could aid in the development of a small-molecule or antibody-based therapy to block this myelosuppressive function of sBARF1, thus allowing more effective immune therapy.
Footnotes
Acknowledgments
The authors are grateful to J.J. Lindenberg for technical and scientific assistance. This project was financially supported by the Dutch Cancer Society Project KWF-VU2007-3776.
Author Disclosure Statement
No competing financial interests exist.