
Abstract
IL-17 producing CD4 T cells have recently been shown to play an important role in mucosal immunity in HIV infection. But its role in peripheral immunity and the molecular mechanism underlying its regulation during HIV-1 infection are ill defined. In this study, we report a significant negative correlation between IL-17 production in peripheral blood and HIV-1 plasma viral load (pVL). On further investigation, we observe a marked reduction in retinoid-related orphan nuclear receptor (RORγt; Th17 lineage specific transcription factor) binding at IL-17 promoter in HIV patients with high viremia (pVL>10,000 copies/mL) in contrast to relatively low viremic patients which indicate the magnitude of viral copy number on RORγt binding at IL-17 promoter. Additionally, our study highlights that FoxP3 influences IL-17 production by binding to and acting together with RORγt, consequently inhibiting RORγt binding to IL-17 promoter with growing viremia in HIV infection. Collectively, our data suggest that FoxP3 interacts with RORγt transcription factor in a viral load-dependent fashion and brings about negative impact on IL-17 production in HIV-1 infection.
Introduction
However, Th17 lineage appears to be involved in the immune system's fight against HIV infection (10,11,14,16,21); it is still unclear what are the molecular determinants involved in its regulation during HIV infection. Therefore, there is an urgent need to understand that how HIV modulate Th17 cells, either directly or indirectly by altering their differentiation program during the immune combat against HIV.
Despite some discrepancies, it has been demonstrated that expression of RORγt is able to induce IL-17 expression in CD4 T cells (2,7,13,18). The transcription factors STAT3 (2,7), FoxP3 (12,26,27), Batf (17), and RunX1 (26) are also involved in controlling the expression of IL-17 cytokine. But limited data regarding the molecular events behind the Th17-mediated HIV immunopathogenesis clearly demonstrate an urgent need to further dissect the immune-dynamics of Th17 cells in HIV-1 pathogenesis. Thus, through this study we tried to explore the HIV-1 subtype ‘C’ infection for defining the molecular determinants involved in Th17-mediated HIV-1 immunopathogenesis further. Hence, we investigated the role of IL-17 producing CD4 T cells in peripheral blood of HIV-1 infected patients at two interdependent “check points”.
The first is associated with the expression of IL-17 cytokine via CD4 T cells, and the second checkpoint involves the regulatory mechanism of IL-17 expression in peripheral blood of HIV individuals. In the light of recent findings, we hypothesize that these two events are largely dependent on the HIV disease progression which in turn is regulated by the HIV plasma viral load. In order to define better the Th17 cellular dynamics and its impact on HIV disease kinetics, we aimed to identify the molecular determinants involved in Th17-based host immunity against HIV-1 subtype ‘C’ infection. Here, we report that HIV-1 viral copy number influences the RORγt DNA binding activity at IL-17 promoter in a FoxP3-dependent manner which ultimately controls the production of IL-17 cytokine in peripheral blood of HIV-1 subtype ‘C’ infection.
Method
Study subjects
The current study was conducted at the HIV National Reference Laboratory (HIV-NRL), Department of Microbiology, All India Institute of Medical Sciences (AIIMS), New Delhi, and Saifia College of Science, Barkatullah University, Bhopal, India. All the patients were recruited at HIV-NRL, AIIMS, New Delhi, after getting the written and informed consent prior to entering the study; the study was approved by the Institutional Ethics Committee. 15 HIV-1 seropositive highly active anti-retroviral therapy (HAART) naive subjects and 5 HIV uninfected healthy control subjects were recruited for this study. Based on plasma viral load, 15 HIV-1 seropositive individuals were characterized in three categories: (1) pVL<47 copies/mL, (2) pVL: 47–10,000 copies/mL, (3) pVL>10,000 copies/mL.
Cell preparation and stimulation
Peripheral blood mononuclear cells (PBMCs) were isolated, counted, and checked for viability. For each patient sample, 1×106 PBMCs were distributed and cultured in RPMI 1640 with L-glutamine (Sigma Aldrich) supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen), anti-CD28 and anti-CD49d (1 μg/mL, BD Biosciences) co-stimulatory antibodies in 96-well round-bottomed plates (Costar, Cambridge, MA) and stimulated with a set of 121 overlapping peptides corresponding to the consensus HIV-1 subtype ‘C’ Gag peptide of 510 amino acids (NIH, USA) (2 μg/mL). Control conditions included stimulation of cells with medium alone as negative control to assess spontaneous production of IL-17 and with PMA (5 ng/mL, Sigma-Aldrich), Ionomycin (1 μM, Sigma-Aldrich)) as positive control to ensure the cells were responsive. For flow cytometric analysis, cultures were incubated for 2 h at 37°C in a 5% CO2 incubator, followed by additional 4 h incubation in the presence of cytokine secretion inhibitor Brefeldin A (1 μg/mL; Sigma-Aldrich) and stained as described (19). For further experiments (Electrophoretic Mobility Shift assay and Western blot analysis) PBMCs were harvested after 6 h of stimulation and Th17 cells were sorted from PBMCs using staining reagents from the IL-17-Secretion Assay-Detection Kit (Miltenyi Biotec). In brief, cells were first labeled with IL-17 catch reagent and incubated for 5 min at 37°C in serum-free medium, followed by another 45-min incubation period on a slow, continuous rocker at 37°C. Finally, cells were stained with an IL-17 PE Antibody (Ab) together with anti-CD4-PercpCy5.5 and anti-CD3-APC-Cy7. Th17 cells were sorted as CD3+CD4+IL-17+ using a BD FACS Aria cell sorter system (BD, Biosciences). Cytoplasmic extract and nuclear extract was prepared from sorted Th17 cells as described (5).
Quantification of plasma HIV-1 RNA
Viral RNA was extracted from all the collected plasma samples of patients and quantified by COBAS TaqMan-48 assay (Roche Diagnostic, Meylan, France) with a lower limit of detection of 47 copies/mL strictly following the manufacture's instruction.
Flow cytometry
The following antibodies used for flow cytometry were purchased from Becton Dickinson (BD) biosciences: APC-Cy7 conjugated CD3, PercpCy5.5 conjugated CD4 and PE conjugated IL-17. For each analysis, 50,000–100,000 events were acquired on a LSR-II flow cytometer (BD Biosciences) and data were analyzed using FlowJo software (Tree Star, CA).
Electrophoretic Mobility Shift Assay (EMSA) and Supershift EMSA
Electrophoretic mobility shift assay (EMSA) was performed as described (20) using following P32-labeled DNA probes containing RORγt binding site derived from human IL-17 promoter (9): −148/-114 region with wild-type RORγt binding sites (WT): 5′CTTCAGAAGGAGAGATTCTTCTA
For supershift analysis, 4 μg of anti-RORγ transcruz antibody (Santa Cruz Biotechnology, Inc.) was added to the nuclear extracts and incubated for 0.5 h at room temperature prior to addition of the P32-labeled DNA probes.
Western blot assay
Western blotting was carried out as described (23). Membranes were probed with antibodies (Abs) specific for RORγt (E-Biosciences) and β-actin (Santa Cruz Biotechnology). Binding of secondary HRP-labeled Abs (Santa Cruz Biotechnology) was analyzed using super signal west pico chemiluminescent substrate (Pierce, Rockland, II).
Co-Immunoprecipitation assay
Co-Immunoprecipitation (Co-IP) assay was performed as described (23). Whole cell lysate prepared from Gag-stimulated Th17 cells, were precleared with protein G-sepharose beads and RORγt was immunoprecipitated using protein G-sepharose beads precoated with αRORγt Ab (E-Biosciences). For pull down experiments, cells were chilled on ice, resuspended in 1 mL of the cell permeable protein crosslinker (dimethyl 3, 3′ diethiopropionimidate dihydrochloride, Sigma Aldrich) in PBS (2 mg/mL), and incubated at room temperature for 20 min. A whole cell lysate was prepared and RORγt immunoprecipitated with αRORγt Ab bound protein G sepharose beads. Western blot was probed with Ab specific for FoxP3 (Santa Cruz Biotechnology, Inc.) and RORγt (E-Biosciences).
Transfection of cells with FoxP3 siRNA
CD3+CD4+ Cells (sorted via FACS Aria) were transfected with FoxP3 siRNAs (Santa Cruz Biotechnology, Inc.), using Lipofectamine LTX (Invitrogen, Carlsbad, CA) as per the manufacturer's instructions. Briefly, cells (3×106cells/well of a 6-well plate) were seeded in 1.5 mL of RPMI medium. The siRNA specific for the human FoxP3 gene to be silenced was added to 500 μL of RPMI medium in Eppendorf tube; 3 μL of PLUS reagent was added to the same tube. The contents of tube were mixed and incubated at RT for 5 min. Subsequently, 13 μL of Lipofectamine was added to the tube. The solution was mixed gently by pipetting up and down and then the tube was kept at RT for 30 min. The contents of the tube were added to cells in plate. The plate was kept at 37°C in a CO2 incubator. After 4 h of transfection, 1.5 mL of RPMI containing 20% FBS was added per well without removing the transfection mixture. After 48 h, the cells were harvested and subjected to Gag stimulation as previously mentioned. Transfection efficiency was determined by transfecting cells with fluorescein conjugated control siRNA, and measuring the frequency of fluorescein positive cells via flow cytometry. Approximately >75% of cells were transfected under the conditions used. Expression of FoxP3 was analyzed by Western blot. Impact of Foxp3 silencing on RORyt mediated IL-17 expression was analyzed by Chromatin Immunoprecipitation Assay (ChIP) and % of IL-17 producing CD4 T cells were estimated on a LSR-II flow cytometer (BD Biosciences).
ChIP assay
Cells (1×107/well) were stimulated with Gag stimulus for 6 h. ChIP was performed using ChIP-IT kit (Active Motif) following the manufacturer's instructions. After immuno-precipitation using anti RORγt antibody (E-Biosciences), followed by DNA extraction, PCR was performed to amplify -353/-32 region of the IL-17 promoter using following primer set: forward 5′CACCAGAAAGACCTACATGTTAC3′ and reverse 5′GGGCTTTCTCCTTCTGTGGTC3′. For a negative control human GAPDH promoter was amplified by using primers: forward 5′CGGGATTGTCTGCCCTAATTAT3′ and reverse 5′GCACGGAAGGTCACGATGT3′.
Results
Reduced production of HIV-1 Gag specific IL-17 producing CD4 T cells in HIV-1 individuals with high plasma viremia
To evaluate the influence of pVL on the expression of IL-17 cytokine via CD4 T cells, 15 ART naive HIV-1 infected patients were categorized: (1) Group-1; pVL<47 copies/mL, (2) Group-2; pVL: 47–10,000 copies/mL, (3) Group-3; pVL>10,000 copies/mL and studied. Clinical characteristics of study cohort are presented in Table 1. To examine the cell's responsiveness, we used PMA-Ionomycin stimulation as positive control and observed induced IL-17 cytokine production in all clinical categories compared to the unstimulated controls ((mean±SD) Group 1, 5.4±5.6; Group 2, 5.5±7.3; Group 3, 0.9±0.4 and control subjects, 1.7±0.6) (Fig. 1A, 1B).
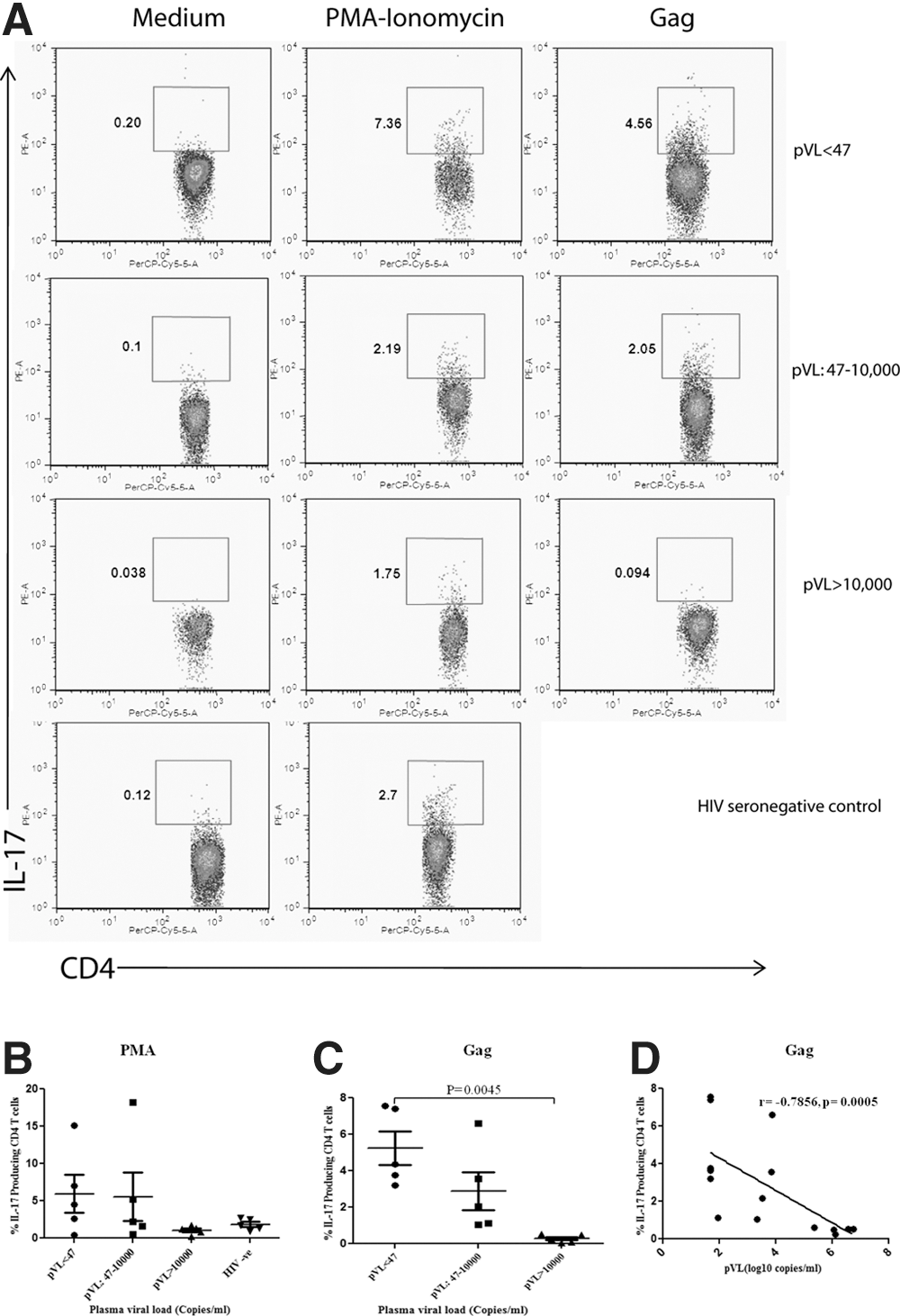
Next, to examine the antigenic specificity, we stimulated PBMCs with HIV-1 subtype ‘C’ Gag pool of peptides ex vivo. Representative data of IL-17 production by CD4 T cells from above mentioned clinical categories and uninfected controls are shown in Figure1A, which illustrates the impact of HIV-1 viremia on the expression of IL-17 cytokine in response to HIV-1 subtype ‘C’ Gag peptide pool. HIV-1 Gag specific % IL-17 producing CD4 T cells were predominantly detected in patients with low viremia and was significantly reduced at relatively high viremia (p<0.05) ((mean±SD): Group 1, 5.11±2.1; Group 2, 2.8±2.3; Group 3, 0.47±0.12) (Fig. 1C). On further analysis, we observed a significant negative correlation (ρ,−0.78; p=0.0005) between HIV-1 pVL and Gag specific % IL-17 producing CD4 T cells (Fig. 1D). Collective analysis strongly suggests that individuals with relatively low pVL established stronger HIV-1 specific IL-17 responses, than those with higher pVL.
Subtype ‘C’ specific Gag induces RORγt DNA binding activity to IL-17 promoter
Subsequently, we wanted to investigate the molecular determinants of IL-17 expression in all the above mentioned clinical categories involved in its regulation. Since RORγt is a key regulator of IL-17 production (2,13,18), thus we speculate involvement of RORγt factor in the regulation of HIV-1 induced IL-17 production. To investigate this possibility, the binding site of RORγt protein on IL-17 promoter was ascertained. An approximately 500 bp DNA sequence upstream of the transcriptional start site of IL-17 gene was selected as the promoter region (9). Sequence analysis revealed the presence of potential RORγt binding element -125TGACCT-120 within the IL-17 promoter, and RORγt-mediated regulation of IL-17 gene expression in HIV-1 infection was investigated. For this purpose, nuclear RORγt DNA binding activity to -125/-120 region of IL-17 promoter, containing potential RORγt binding elements, were determined via EMSA using oligonucleotides specific for the respective region as probes in the previously described clinical categories of HIV-1 infection in response to both HIV-1 specific Gag stimulus and nonspecific PMA-Ionomycin stimulus. Representative figures of RORγt binding to IL-17 promoter from each clinical categories of the study cohort is illustrated in Figure 2A.
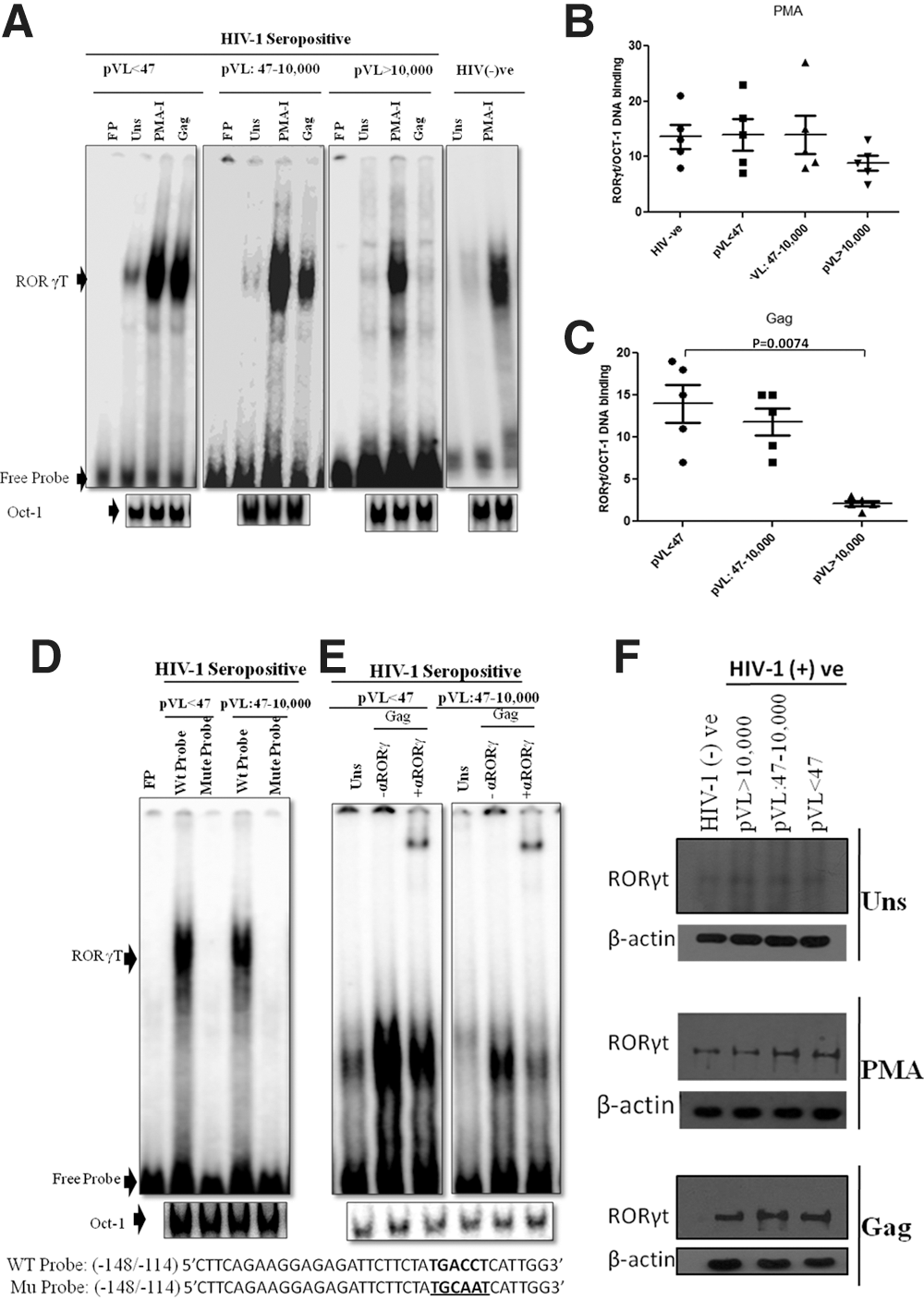
In response to the PMA-Ionomycin stimulus, we observed an (5–27 fold) increase in RORγt DNA binding activity in all clinical groups including HIV seronegative control samples when compared to unstimulated controls, whereas on statistical intergroup analysis we could not observe any significant differences in RORγt DNA binding activity among all clinical categories (Fig. 2B). Furthermore, relative to untreated cells, a 1–19-fold increase in RORγt DNA binding activity was observed in Gag-treated cells. Moreover, intergroup analysis revealed a significant (p=0.0074) reduction in RORγt DNA binding to IL-17 promoter in Group 3 (pVL>10,000) patients when compared to Group 1 (pVL<47) patients (Fig. 2C). This reveals that reduction in RORγt DNA binding activity is Gag specific. Therefore, only Gag stimulus was selected as “reference” for further study.
To exclude the possibility of any nonspecific DNA binding in patients with pVL<10,000 copies/mL, we assayed RORγt DNA binding activity using oligonucleotide -148/-114 IL-17 sequences with mutated RORγt binding site, which failed to exhibit any nuclear DNA binding activity in both categories (Fig. 2D). In addition, using transcruz antibody specific for human RORγ, supershift analysis further confirmed the nuclear RORγ DNA binding activity to the probe specific for -148/-114 regions of IL-17 promoter in Gag-stimulated cells (Fig. 2E).
RORγt protein expression remains unaltered during the course of HIV-1 infection
In contrast to our EMSA data, RORγt expression remained constant among previously mentioned clinical categories in response to both PMA-Ionomycin and Gag stimulus during the course of HIV-1 infection; also in comparison the level of β-actin protein was remained constant, which revealed that HIV-1 infection only impinge on the DNA binding ability of RORγt protein, not on its expression (Fig. 2F).
FoxP3 reduces the nuclear DNA binding activity of RORγt to IL-17 promoter in the course of HIV-1 infection
Next, we wanted to identify what other factors are involved in this regulatory mechanism. Therefore, we carried out a Co-IP assay to discern the other putative protein components of this RORγt protein complex. Several groups have demonstrated that FoxP3 protein participates in the regulation of RORγt mediated IL-17 protein expression (12,26,27). Therefore, the possibility that FoxP3 protein involved in RORγt mediated IL17 expression was investigated using the Co-IP assay in previously described clinical categories of HIV-1 infection. In contrast to EMSA data, we observed lack of FoxP3 protein bound to RORγt in Group 1 (pVL<47 copies/mL) HIV-1 patients, but this binding was progressively induced in Group 2 (pVL: 47–10,000 copies/mL) and Group 3 (pVL>10,000 copies/ml) HIV-1 patients (Fig. 3A). This observation gave us a clue that FoxP3 might be involved in repression of RORγt-mediated IL-17 production with rising plasma viral load in HIV-1 infection.
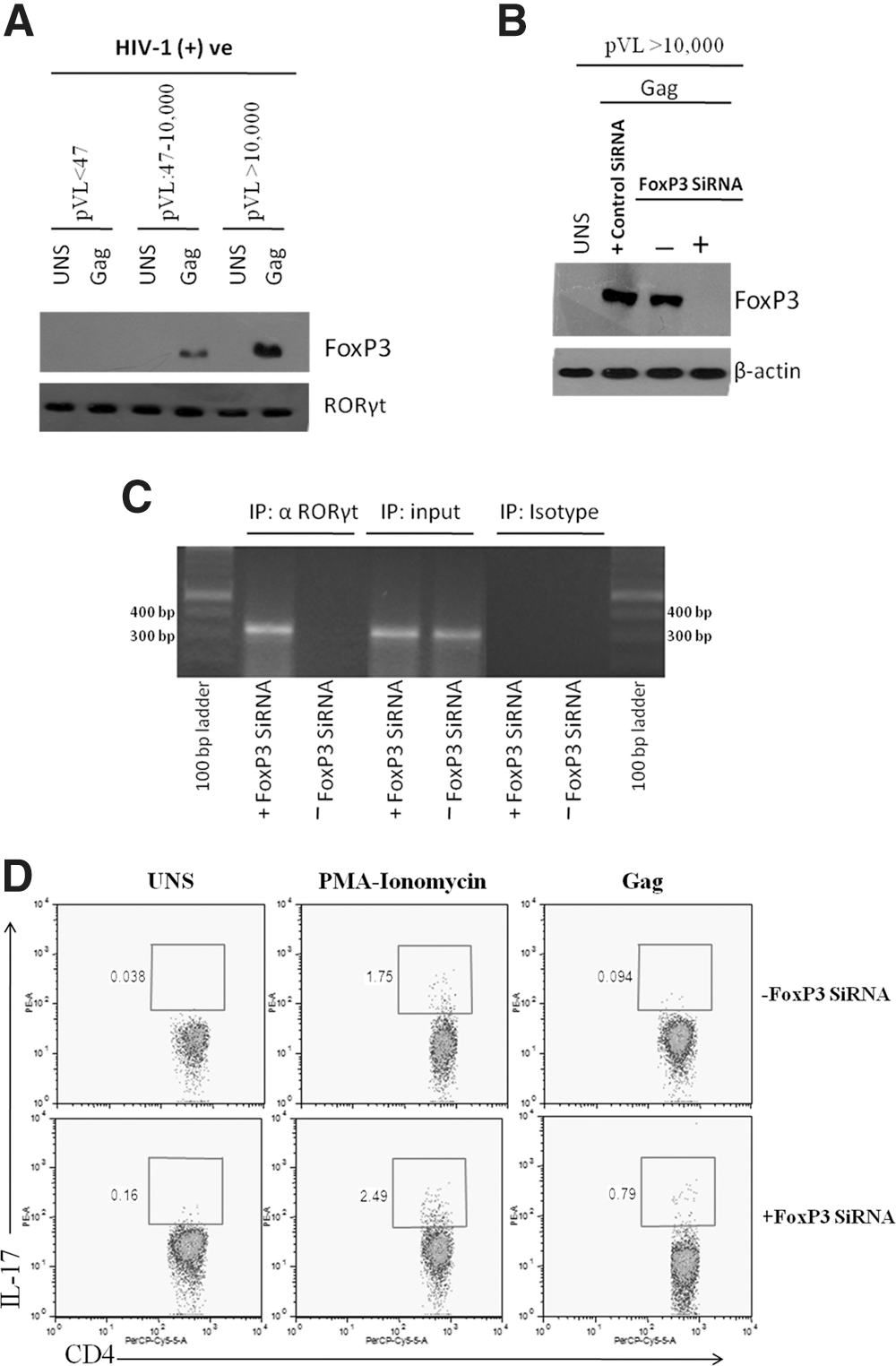
FoxP3 is associated with RORγt and inhibits IL-17 production via CD4 T cells in HIV-1 infection at high viral load.
Therefore, to further verify the negative impact of FoxP3 expression on the RORγt-dependent IL-17 production, FoxP3 gene was silenced in Group 3 (pVL>10,000 copies/mL) patients where FoxP3 was virtually overexpressed, but RORγt mediated IL-17 expression was relatively diminished (Fig. 3B). ChIP analysis using an anti-RORγt Ab was then performed to determine whether FoxP3 silencing may induce Gag-dependent RORγt binding to the IL-17 promoter at high viremia. The data presented in Figure 3C, indicate that dearth of FoxP3 releases RORγt so that it can bind directly to putative RORγt binding sites in the human IL-17 promoter. In particular, RORγt which bound at the IL-17 promoter could be inhibited by FoxP3 transcription factor at high viremia in HIV-1 infection.
On further analysis, we reported that in contrast to unsilenced control samples, FoxP3 gene silencing contrived % IL-17 producing CD4 T cells by 3–8-fold and 1.4–3-fold induction in Group 3 patients responding to Gag and PMA-Ionomycin stimulus, respectively. Anticipated from the ChIP experiment, we found that FoxP3 silencing leads to improved IL-17 production when compared to parallel control. Hence, it is tempting to speculate that high HIV-1 plasma viremia-induced FoxP3 protein, subsequently detains RORγt transcription factor and prevents its binding to IL-17 promoter required for switching on the IL-17 production machinery.
Discussion
In the current study, we report a significant negative correlation between the frequency of IL-17 producing CD4 T cells and HIV-1 plasma viremia. Similarly, enhanced IL-17 production was reported in HIV-1 infected children with a pVL of below 50 copies/mL in contrast to those with detectable HIV (21). Also, HIV-1-specific and CMV-specific IL-17 producing CD4 T cells were detectable in early HIV-1 infection but were undetectable in chronic and nonprogressive HIV-1 infection (25). As well, an increased number of Th17 cells in long-term nonprogressors was also reported by another group (22). These findings, together with our finding, strongly suggest that IL-17-producing T cells are associated with viral load and disease progression. This also indicates that improved production of IL-17 cytokine-producing CD4 T cells could lead to more preserved immune response against opportunistic infections, thereby leading to reduced immune activation and could explain the slower disease progression. This reinforces the notion that a possible functional impairment of Th17 cells in patients with detectable HIV-1 levels, and that restoration of Th17 cells, could be possible through suppression of viremia.
Although it is clear from our study and above mentioned evidence that IL-17-producing CD4 T cells are important in HIV immunopathogenesis, but it is still unclear how IL-17 production is regulated during HIV infection. Understanding how HIV modulates Th17 cells is crucial to restoring its population and function, and can enable us to design treatments strategies to increase Th17 levels, beneficial to HIV infection.
Studies of the transcription of genes encoding Th1/Th2 cytokines have shown that such transcription is controlled by cis-regulatory elements evolutionary conserved and located in the vicinity of gene encoding the cytokine (1 –3,7,26). Therefore, we made an attempt in this direction and performed a search for potential regulatory element within the IL-17 locus and identified one such evolutionarily conserved ROR response element (RORE), present within 200 bp IL-17 promoter region. RORE consist of the consensus core motif AGGTCA preceded by a 5 bp A/T rich sequence (1,2,26). Since RORE is recognized as RORγt binding site, suggesting that transcription of IL-17 may be directly regulated by RORγt transcription factor. (7,18).
In view of the above, we investigated the role of RORγt in IL-17 transcription. Responding to Gag stimulus, induced RORγt binding to RORE at IL-17 promoter in HIV-1 patients with undetectable plasma viremia was observed in comparison to HIV-1 patients with relatively high plasma viremia. This circuitously indicates a negative correlation between RORγt binding activity at IL-17 promoter and HIV-1 plasma viremia. Therefore, we propose that the gradual loss of IL-17 production during the course of HIV-1 infection may possibly be regulated by the RORγt transcription factor. This conclusion was supported by various results demonstrating that RORγt is directly involved in IL-17 transcription (13,18,26). This finding also suggests that intracellular viral copy number plays an important role in IL-17 production during course of HIV-1 infection.
Furthermore, another possibility for the gradual reduction in RORγt nuclear binding ability could be related to its protein expression. Therefore, we investigated RORγt protein expression in both Gag and PMA-Ionomycin stimulated cells. Another important finding arising from this study is that RORγt protein expression remains constant in all clinical categories, including control subjects, in response to both Gag and PMA-Ionomycin stimulation. Therefore, we concluded that although Gag stimulation does not induce concomitant degradation of RORγt protein but indeed brings on reduction in DNA binding activity of RORγt at IL-17 promoter in HIV-1 patients with relatively high viral load when compared to HIV-1 individuals with undetectable viral load. Hence, viral copy number encroaches on RORγt DNA binding activity, not on its expression, and influences IL-17 production in HIV-1 infection.
However, responding to nonspecific PMA-Ionomycin stimulus we observed virtually unaltered RORγt binding at IL-17 promoter in intergroup analysis of study subjects. This shows that reduction in RORγt DNA binding activity is HIV-1 specific. Therefore, only Gag stimulus was selected as “reference” for further study to analyze the basis of alteration in RORγt binding activity at IL-17 promoter in the course of HIV-1 infection.
Additionally, it has been well documented by several groups that FoxP3 also influence IL-17 production (12,26,27). In this regard, Zhou et al (27) observed that in vitro TGF-β induced FoxP3, inhibits Th17 differentiation by antagonizing RORγt function, at least in part through their interaction. This evidence prompted us to investigate the FoxP3 interaction with RORγt in the regulation of IL-17 production during the course of HIV-1 infection. Importantly, we observed that FoxP3 physically interacts with RORγt, in the repression of RORγt-induced IL-17 production at high viremia (pVL>10,000 copies/mL). This finding also implies that reduced expression of FoxP3 was inversely correlated with RORγt binding at IL-17 promoter. Conversely, Zhou et al., (27) observed that many of the IL-17+ cells also expressed FoxP3, thus RORγt-dependent IL-17 expression can occur in the presence of FoxP3, but the level of FoxP3 may be insufficient to block RORγt function or alternatively, IL-6 may overcome the inhibitory function of FoxP3.
As an effort to determine whether FoxP3 suppression may normalize the RORγt binding to IL-17 promoter in HIV-1 infection, FoxP3 silencing was followed by a ChIP assay using αRORγt ab in the third category patients (pVL>10,000 copies/mL). Results obtained using αRORγt ab in ChIP assay showed that inhibitory effect of FoxP3 protein on RORγt binding to IL-17 promoter at high viremia can be controlled at some extent. Furthermore, a relative induction in IL-17 production via CD4 T cells after FoxP3 silencing was observed when compared to corresponding control samples. On the other hand, when we compared the induced production of % IL-17 producing CD4 T cells in Group 3 patients (after FoxP3 silencing) with Group 1 patients, we could not observe comparative data. This suggests that beside FoxP3 and RORyt, IL-17 production could be controlled by other regulatory elements. Functional impairment of RORyt at high viremia could be another valid possibility behind the failure in complete restoration of IL-17 production in HIV-1 infection, even after FoxP3 elimination. Hence, our study emphasizes the notion that HIV-1 viral load induced, FoxP3 regulated RORyt-mediated IL-17 production is “multifactorial” which demands further dissection.
This ‘yin-yang’ relationship of RORγt and FoxP3 is the probable basis of the observation that the differentiation of Th17 cells and Treg cells is often reciprocal (11,12,14,26,27). Besides, recent reports indicate that HIV disease progression is associated with loss of Th17/Treg relative balance (14). The decision of antigen stimulated cells to differentiate into either Th17 or Treg cells depend on the cytokine regulated balance of RORγt and FoxP3 (27). Therefore, collectively this evidence, along with our observation, specifies that relative balance of RORγt and FoxP3 is the key to the HIV disease progression.
Importantly, if we dissect the clinical characteristics of all the study subjects, co-infections (Table 1) present in the third group of patients (pVL>10,000 copies/mL) could contribute to skew Th17 cell differentiation program leading to inhibition of IL-17 expression in comparison to patients of Group 1 and Group 2; however, the real bearing of this observation in HIV-1 infection needs further detailed investigations.
A very valid limitation of this data is the vast gap between in vitro and in vivo systems. The in vitro experiments provide us accurate quantitative data on promoter binding specificities, but in cells, binding occurs in the context of correct complement of other regulatory proteins and post translational modifications. Therefore, new and more sensitive techniques are required to generate and reproduce the data. Also, whether RORγt activity is regulated by a ligand in HIV-1 infection is not yet known. Thus another important caveat of this study is to identify the specific natural ligand involved in this regulatory mechanism.
In conclusion, our data suggest that at high viral load (pVL>10,000 copies/mL), FoxP3 protein interacted with RORγt transcription factor which possibly induces conformational changes in RORγt protein in such a way to inhibit its DNA binding ability at IL-17 promoter without disturbing its expression and switching off the transcription machinery of IL-17 cytokine.
The identification of RORγt and FoxP3 as key regulators of the Th17 lineage in HIV-1 infection suggests that targeting these molecules could be a viable strategy for treating HIV-associated Th17 effector functions. It is tempting to speculate that manipulation of RORγt activity may be an attractive therapeutic strategy in HIV-1 infection.
Footnotes
Acknowledgments
We would like to thank Dr. Pradip Sen (Institute of Microbial Technology, Chandigarh) who helped us in FoxP3 gene silencing and ChIP assay experiments. Finally, we thank the Indian Council of Medical Research (ICMR), National AIDS Control Organization (NACO) and AIIMS for providing the research infrastructure and financial support. This research work was funded by Institutional Research Committee of All India Institute of Medical Sciences (AIIMS), New Delhi.
Author Disclosure Statement
The authors have no potential conflict of interest related to this article.