
Abstract
Myeloid dendritic cells (mDCs) are the most potent professional antigen-presenting cells that regulate specific T-cell responses. Here we studied the ability of mDCs to kill T cells during HCV infection. We found that mDCs from chronic hepatitis C (CHC) patients expressed upregulated levels of two inhibitory ligands, Fas ligand and the ligand 2 of PD-1 (PD-L2), compared to healthy mDCs. However, their expression of the ligand 1 of PD-1 (PD-L1), tumor necrosis factor-related apoptosis inducing ligand (TRAIL), and B lymphocyte stimulator (BLyS) on the cell surface was comparable to healthy mDCs. CHC patient mDCs had cytotoxic effects on autologous patient T cells and allogeneic healthy T cells. CHC patient T cells had increased expression of PD-1 compared to healthy T cells. These results indicate that the cytotoxic activity of mDCs is upregulated to kill T cells during chronic HCV infection, which represents a novel mechanism of HCV immune evasion.
Introduction
Dendritic cells (DCs) are the most potent professional antigen-presenting cells (APCs) that regulate T-cell responses (8,9). Besides stimulating T-cell proliferation, DCs also have the ability to induce immune tolerance by killing T cells. Killer DCs express inhibitory molecules, including Fas ligand (FasL: CD178) (10 –16) and tumor necrosis factor (TNF)-related apoptosis inducing ligand (TRAIL: CD253) (17 –20). Recently, the presence of killer DCs has been reported during measles virus (MV) (17,18) and HIV (19,20) infections. MV induces TRAIL expression in DCs, which then kill activated T cells (17) and tumor cells (18). During HIV infection, killer DCs produce TRAIL and induce the apoptosis of CD4 T cell lines (19,20).
As for HCV infection, there was only one report by Ciesek et al. (21) examining the killing activity of myeloid DCs (mDCs). The study by Ciesek et al. showed that mDCs from HCV-infected patients do not increase TRAIL production and can not kill T cells, and thus the authors concluded that patient mDCs displayed no cytotoxic activity (21). However, the study by Ciesek et al. used immortalized leukemia cell lines as target cells in killing assays, which might not be best representative of T cells during HCV infection. Furthermore, the study by Ciesek et al. only examined the expression of TRAIL molecules on mDCs, while the expression of other inhibitory molecules remains to be determined. In addition to the unknown expression of FasL on mDCs during HCV infection, programmed death-1 (PD-1) ligands 1 and 2 (PD-L1:CD274 and PD-L2:CD273) are also inhibitory molecules that can induce T cell apoptosis (22), but their roles during HCV infection have yet to be studied. In brief, whether DCs kill T cells during HCV infection requires further study.
Our previous study (23) showed that the ability of mDCs to stimulate CD4 T-cell proliferation was impaired in CHC patients. We previously found that mDCs in CHC patients have decreased expression of HLA-DR and CD86. However, the expression of a novel co-stimulatory molecule, B lymphocyte stimulator (BLyS), on mDCs during HCV infection has not been reported. BLyS is a member of the tumor necrosis factor (TNF) superfamily and is also termed as B cell-activating factor belonging to the TNF family (BAFF) (24 –26). Full-length BLyS is expressed on the plasma membrane as membrane-bound BLyS (mBLyS), which is capable of co-stimulating T cell activation (27). mBLyS expressed on DCs has co-stimulatory activity on both CD4+ and CD8+ T cells at multiple stages, including naïve T cells, recently antigen-primed T cells, and memory T cells (27,28).The expression of mBLyS on mDCs has yet to be examined.
In this study, the cytotoxic activity of mDCs from CHC patients was examined. Due to the low numbers of circulating mDCs in peripheral blood, assays were first performed on in vitro generated monocyte-derived mDCs (moDCs), which were monocytes that were cultured in vitro with IL-4 and GM-CSF. The results were then confirmed by performing assays on ex vivo mDCs, which were isolated from peripheral blood using magnetic activated cell sorting (MACS).
The phenotypic assays were performed first, with the expression of inhibitory ligands, including FasL, PD-L1, PD-L2, and TRAIL on moDCs being examined by flow cytometry. The expression of a novel co-stimulatory molecule, BLyS, was also analyzed. Second, killing assays were performed using different target T cells, including T cells freshly isolated from peripheral blood of CHC patients and healthy donors, and the Jurkat T-cell line. The moDCs from CHC patients and healthy donors were co-cultured with target T cells and cell apoptosis was examined by TUNEL. These assays found that patient moDCs have upregulated cytotoxic activity and are capable of killing CD4 and CD8 T cells isolated from human peripheral blood. Third, possible mechanisms for the killing were explored by examining the expression of receptors, Fas and PD-1, on target T cells. Furthermore, to determine whether the killing was mediated by soluble factors or cell–cell contact, moDCs and T cells were cultured either in the presence of blocking antibodies, or in two chambers separated by a polycarbonate membrane in transwells. Finally, mDCs were ex vivo isolated from the peripheral blood, and phenotypic and killing assays were performed to determine if these mDCs of CHC patients demonstrate upregulated cytotoxic activity compared to those of healthy donors.
Patients and Methods
Blood samples
Male CHC patients who had been HCV-RNA positive and had detectable HCV antibodies for more than 3 years were included in this study. If patients were given a trial of therapy with pegylated interferon and ribavirin, they were off therapy for at least 6 months. Patient characteristics are shown in Table 1. Control samples were collected from HCV-negative healthy male donors (age-matched). Ethics approval for human blood collection was obtained from the University of Alberta, Faculty of Medicine Research Ethics Board. Informed consent was obtained from all donors.
In the pilot study, we observed more cytotoxic activity of moDCs from male CHC patients, however weaker cytotoxic activity of moDCs from female CHC patients. As a result, we concentrated on immune cells from males in this study.
Antibodies
Mouse anti-human FasL, PD-L1, PD-L2, TCR, Fas, PD-1, CD11c, and isotype antibodies were purchased from BD Biosciences (San Diego, CA). Alexa 568 conjugated goat anti-mouse IgG was purchased from Molecular Probes (Invitrogen, Carlsbad, CA).
Culture of in vitro generated moDCs and ex vivo isolated mDCs
PBMCs were isolated from fresh heparinized blood by Ficoll-Hypaque density gradient centrifugation. The cells were plated to a density of 3.0×107 cells/well in 6-well plates, incubated for 2 h, and the nonadherent cells were removed. The adherent cells were cultured in RPMI-1640 containing 1% autologous human serum, 50 ng/mL of human GM-CSF, and 10 ng/mL of human IL-4. On the fifth day of culture, 0.1 μg/mL of lipopolysaccharide (LPS, MO) was added to stimulate the moDCs. After 6 h, the cells were harvested (29,30) and>99% were identified as mature moDCs based on cell surface expression of HLA-DR and CD11c.
Ex vivo mDCs were isolated using magnetic activated cell sorting (MACS). In brief, ex vivo mDCs were isolated from PBMCs with >90% purity using the BDCA-1 positive isolation kit (Miltenyi Biotech, CA). The ex vivo mDCs were cultured in RPMI-1640 containing 1% autologous human serum. On the third day of culture, 0.1 μg/mL of LPS was added to stimulate mDCs. After 24 h, the cells were harvested as mature ex vivo mDCs (31).
Culture of primary CD4 T cells and Jurkat T cell line
Primary CD4 T cells were isolated from PBMCs with >95% purity by MACS. Briefly, PBMCs isolated from donors were incubated with FcR blocking reagent and PE-conjugated anti-human CD4-antibody (BD Biosciences) for 30 min at 4°C, washed twice with cold MACS buffer (PBS containing 2% BSA and 2 mM EDTA), and incubated with anti-PE magnetic microbeads (Miltenyi Biotec) for 15 min at 4°C. The cells were washed twice with cold MACS buffer and isolated on magnetic columns (Miltenyi Biotec) according to the manufacturer's instructions (32). T cells were suspended in AIM-V medium (Invitrogen).
A T cell leukemia cell line, Jurkat T cells, was cultured in RPMI 1640 (Invitrogen) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 0.1 mg/mL streptomycin (Sigma-Aldrich) (14).
Flow cytometry analysis of FasL, PD-L1, PD-L2, TRAIL, and BLyS expression on cell surface of moDCs
To block nonspecific binding of antibodies, moDCs (1×107 cells) were incubated with 20 μL of FcR blocking reagent (Miltenyi Biotec) for 15 min. After FcR blocking, moDCs (1×106/sample) were individually incubated with PE-conjugated mouse anti-human antibodies against FasL (20 μL), PD-L1 (20 μL), PD-L2 (20 μL), TRAIL (20 μL), and BLyS (20 μL) for 30 min. Cells were washed twice with 2% FCS in PBS, fixed in 4% paraformaldehyde, and then analyzed by flow cytometry using CellQuest software (33).
ELISA of TRAIL concentration in serum and moDC culture supernatant
ELISA of TRAIL concentration was performed as described in the TRAIL ELISA kit (Diaclone, France). In brief, samples of sera or moDC culture supernatant were added into the wells of the 96-well plate. Biotinylated anti-TRAIL antibody (50 μL/well) was added to each well. The wells were incubated for 3 h at room temperature. After the wells were washed, streptavidin-HRP solution (100 μL/well) was added to each well and incubated for 30 min. After the wells were washed, substrate solution (tetramethybenzidine) was added to each well and incubated for 30 min. After stop solution (H2SO4, 100 μL/well) was added to inactivate the enzyme, absorbance was read on a spectrophotometer at 450 nm wavelength.
Cell apoptosis assay
DCs were washed and re-plated in AIM-V medium in 6-well plates (1×106/well). Primary T cells isolated from human blood or Jurkat T cells were added in 6-well plates at different DC: T-cell ratios ranging from 8:1, 4:1, 2:1, 1:1, 1:2, to 1:4. After 5 h incubation, the cells were collected and fixed with 4% paraformaldehyde. Previous study (14) had shown that 5 h is the optimal co-culture time for killing. Our study confirmed this observation.
To detect apoptosis, the cells were stained with TUNEL using the In Situ Cell Death Detection Kit (Roche, Germany), and analyzed by flow cytometry or confocal microscopy (14,34). TUNEL staining was used to detect nuclear DNA fragmentation, which is a marker of late stage apoptosis.
To observe whether cells in co-culture were DCs and T cells, antibody staining was used in our pilot studies. However, antibody staining was not efficient in labeling cells in the killing assays, because apoptotic cells tend to lose their cell surface markers. Since carboxyfluoresce in diacetate succinimidylester (CFSE) fluorescence is stably retained in both healthy and apoptotic DCs, CFSE was used to label DCs in the killing assay. DCs were suspended in PBS at a concentration of 1×106 cells/mL and incubated with 10 μM of CFSE (Molecular Probes) for 10 min. The DCs were then washed twice with 45 mL of ice-cold culture medium AIM-V before co-culture.
Flow cytometry of TUNEL staining
Cells (2×106) were fixed with 4% fresh paraformaldehyde in PBS (300 μL) for 30 min. Cells were washed with 2% FCS in PBS (4 mL). After permeabilization with 100 μL permeabilization solution (0.1% sodium citrate and 0.1% Triton X-100) for 2 min on ice, cells were washed with 2% FCS in PBS (4 mL). Cells were incubated with 45 μL of TUNEL label solution and 5 μL of enzyme solution for 60 min at 37°C in a humidified atmosphere in the dark. After washing, the apoptosis rate of the DCs was determined by flow cytometry.
Confocal microscopy of TUNEL staining
Confocal microscopy was used to assess mDC morphology and nuclear DNA fragmentation. DCs were cultured on coverslips in 12-well plates and T cells were added to the plates. After co-culture, cells were fixed, permeabilized, and stained with TUNEL, as described previously. The coverslips were secured on slides using a mounting medium containing DAPI (Sigma-Aldrich) to stain the nucleus. Cells were observed using a confocal microscope (LSM510, Carl Zeiss, Germany).
Flow cytometry analysis for expression of Fas, PD-1, and T cell receptor (TCR) on T cells
Primary T cells or Jurkat T cells (2×106 cells/sample) were individually incubated with PE-conjugated mouse anti-human antibodies against Fas (5 μL), PD-1 (20 μL), or TCR (10 μL) for 30 min. Cells were analyzed by flow cytometry (33).
Blocking assays
To determine if the cytotoxic activity of moDCs requires cell-to-cell contact, transwell cell culture inserts (Corning Inc., Corning, NY) were used. T cells and moDCs were separated by a transwell insert (0.4 μm pore) (35) and cultured for 5 h.
To determine which molecules are involved in the cytotoxic activity of moDCs, cells were pre-incubated with individual antibodies (endotoxin-free, 2 μg/mL) against FasL, Fas, PD-L2, PD-1, or TCR for 45 min (1,33,36). The moDCs and target T cells were co-incubated for 5 h and stained with TUNEL. Cell apoptosis was analyzed by flow cytometry.
Results
CHC patient moDCs demonstrate increased expression of FasL and PD-L2 compared to healthy donor moDCs
CHC patient moDCs demonstrated increased expression of FasL (Fig. 1) and PD-L2 (Fig. 2), while their expression of PD-L1 was unchanged compared to healthy donor moDCs (Fig. 3A–B). The expressions of membrane-bound TRAIL (Fig. 3C) and BLyS (Fig. 3D) on the cell surface membrane of moDCs were comparable in CHC patients and healthy donors. The concentrations of soluble TRAIL (sTRAIL) in patient sera (Fig. 3E) and in culture supernatants of patient moDCs (Fig. 3F) were comparable to those of healthy donors.
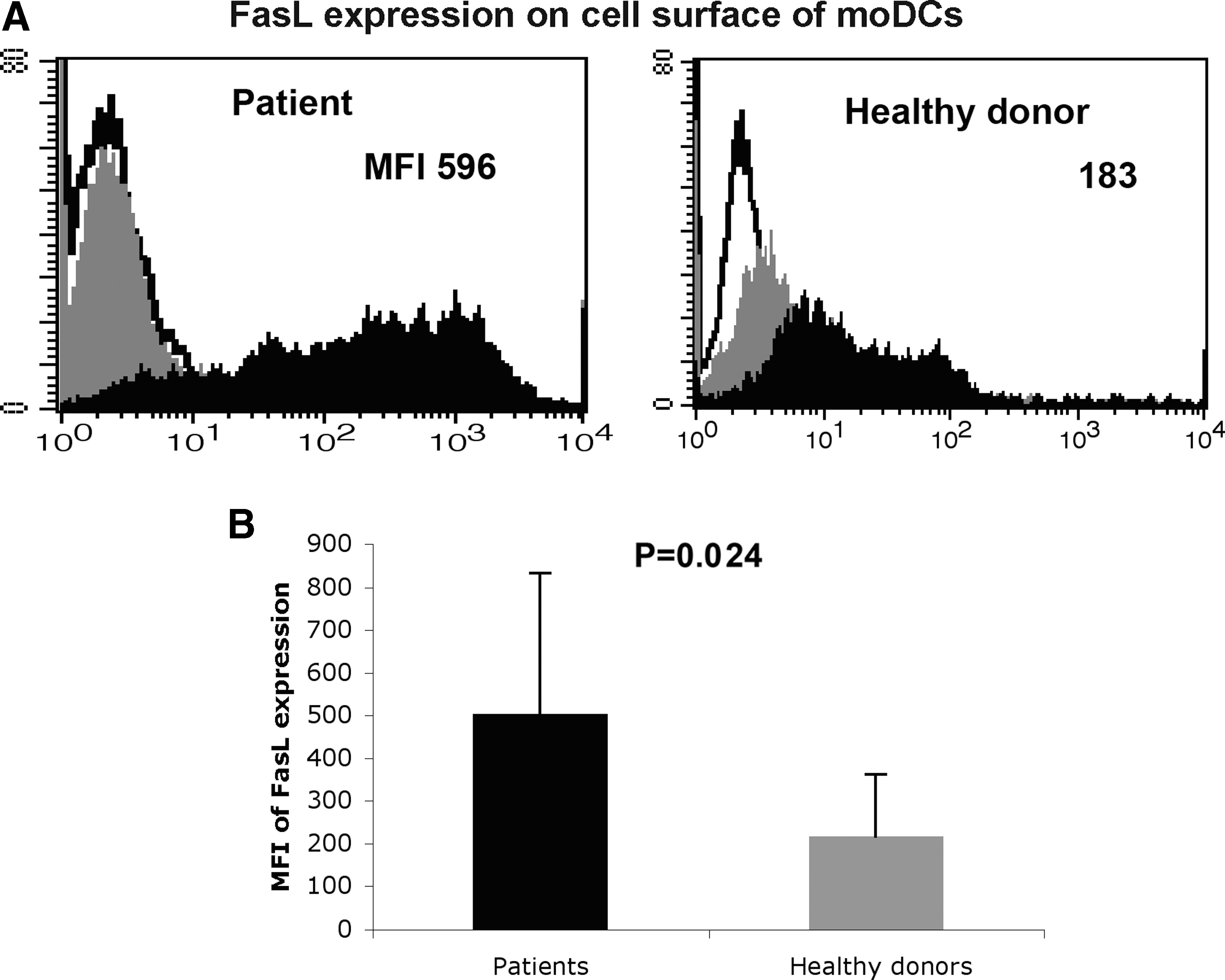
moDCs from CHC patients express FasL at a higher level than moDCs from healthy donors. moDCs were prepared from adherent cells of PBMCs as described in Materials and Methods. The expression of FasL on the cell surface of moDCs from 20 CHC patients and 20 healthy donors was determined. moDCs were stained with specific antibodies or isotype antibodies, and analyzed by flow cytometry. White indicates unstained control, gray indicates isotype control, and black indicates specific staining. Numbers indicate the mean fluorescence intensity (MFI) of FasL expression
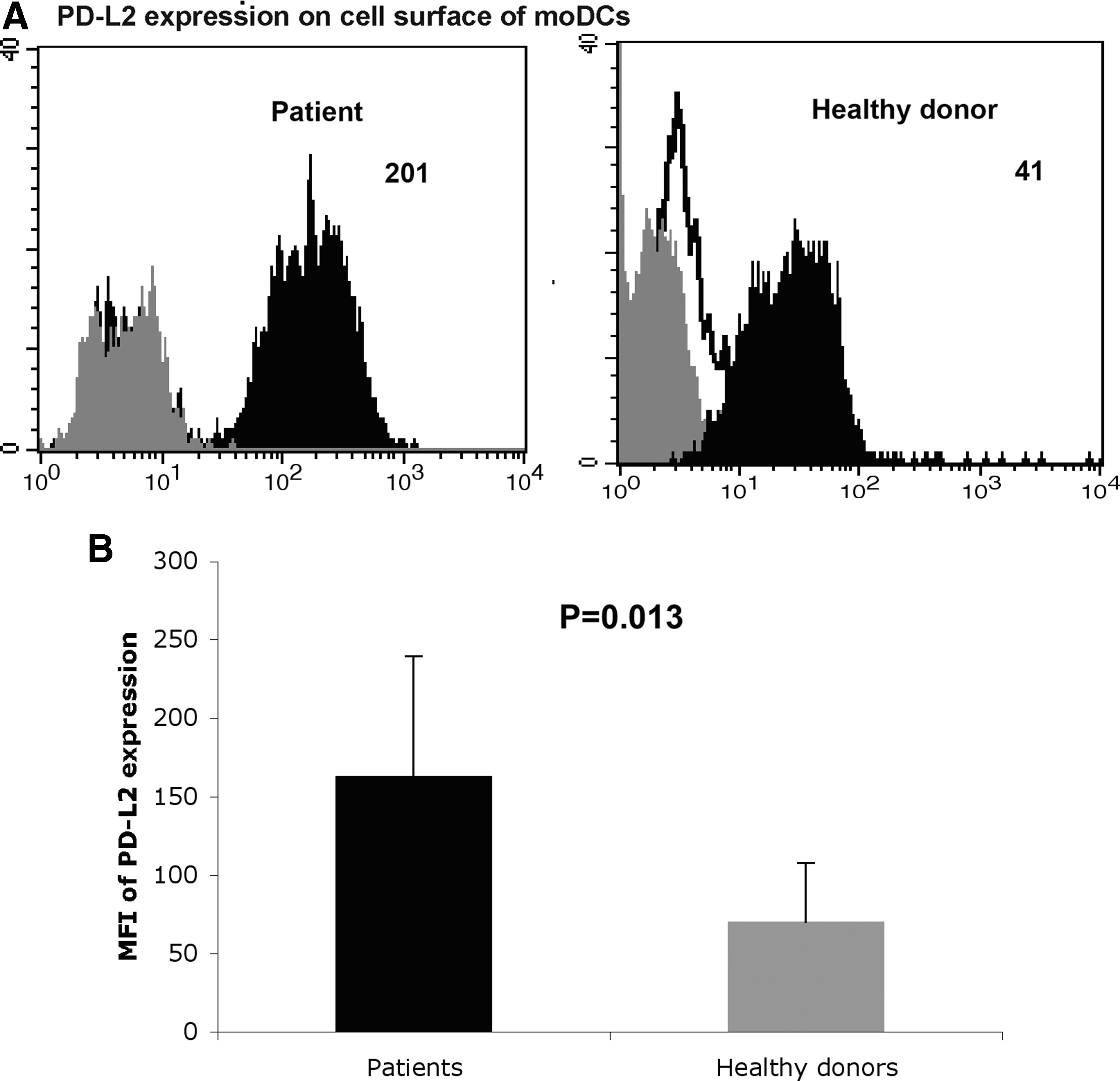
moDCs from CHC patients express PD-L2 at a higher level than moDCs from healthy donors. moDCs were prepared from adherent cells of PBMCs as described in Materials and Methods. The expression of PD-L2 on the cell surface of moDCs from 20 CHC patients and 20 healthy donors was determined. moDCs were stained with specific antibodies or isotype antibodies, and analyzed by flow cytometry. White indicates unstained control, gray indicates isotype control, and black indicates specific staining. Numbers indicate the MFI of specific expressions
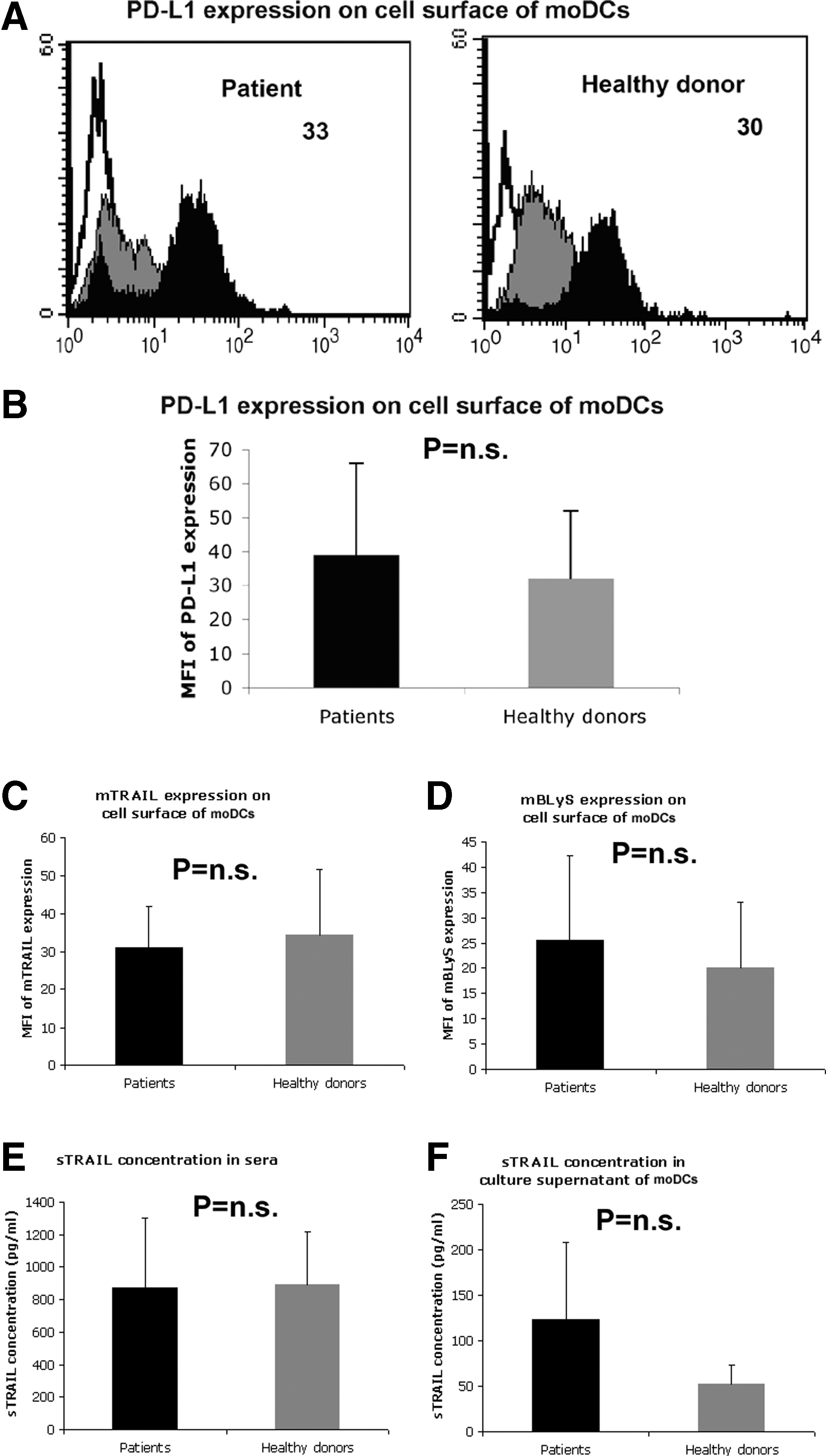
Comparable expression of PD-L1, TRAIL, and BLyS on cell surface of moDCs from CHC patients and healthy donors. The expression of PD-L1, TRAIL, and BLyS on the cell surface of moDCs from 20 CHC patients and 20 healthy donors was determined. moDCs were stained with specific antibodies or isotype antibodies, and analyzed by flow cytometry. White indicates unstained control, gray indicates isotype control, and black indicates specific staining. Numbers indicate the MFI of PD-L1 expression
Increased cell apoptosis in the co-culture of patient moDCs with T cells compared to the co-culture of healthy moDCs with T cells
Since cell death could be apoptosis or necrosis, we used TUNEL staining to identify apoptotic cells.The cell apoptosis percentage was significantly increased when moDCs from CHC patients were co-cultured with healthy CD4 T cells compared to when moDCs from healthy controls were co-cultured with healthy CD4 T cells. This indicated that CHC patient moDCs were cytotoxic to healthy CD4 T cells. The optimal killing was observed at the moDC: T-cell ratio of 4:1 (Fig. 4).

Increased cell apoptosis in the co-culture of patient moDCs with T cells compared to the co-culture of healthy moDCs with T cells. In vitro-generated moDCs from CHC patients and healthy donors were incubated with allogeneic CD4 T cells from a third-party healthy donor. The cells were co-cultured in AIM-V medium for 5 h.
Both healthy CD4 T cells and CHC patient moDCs demonstrate more apoptosis after co-culture
To determine if the increased apoptosis in co-culture was due to moDCs and/or CD4 T cells, CFSE was used to label CHC patient moDCs before co-culture. CFSE+ moDCs and CFSE- CD4 T cells were individually gated by flow cytometry. Interestingly, CD4 T cells demonstrated lower forward scatter (FSC) value (which indicates cell size) after co-culture (Fig. 5A). This suggests cell shrinkage, which is one of the characteristics of apoptotic cells.
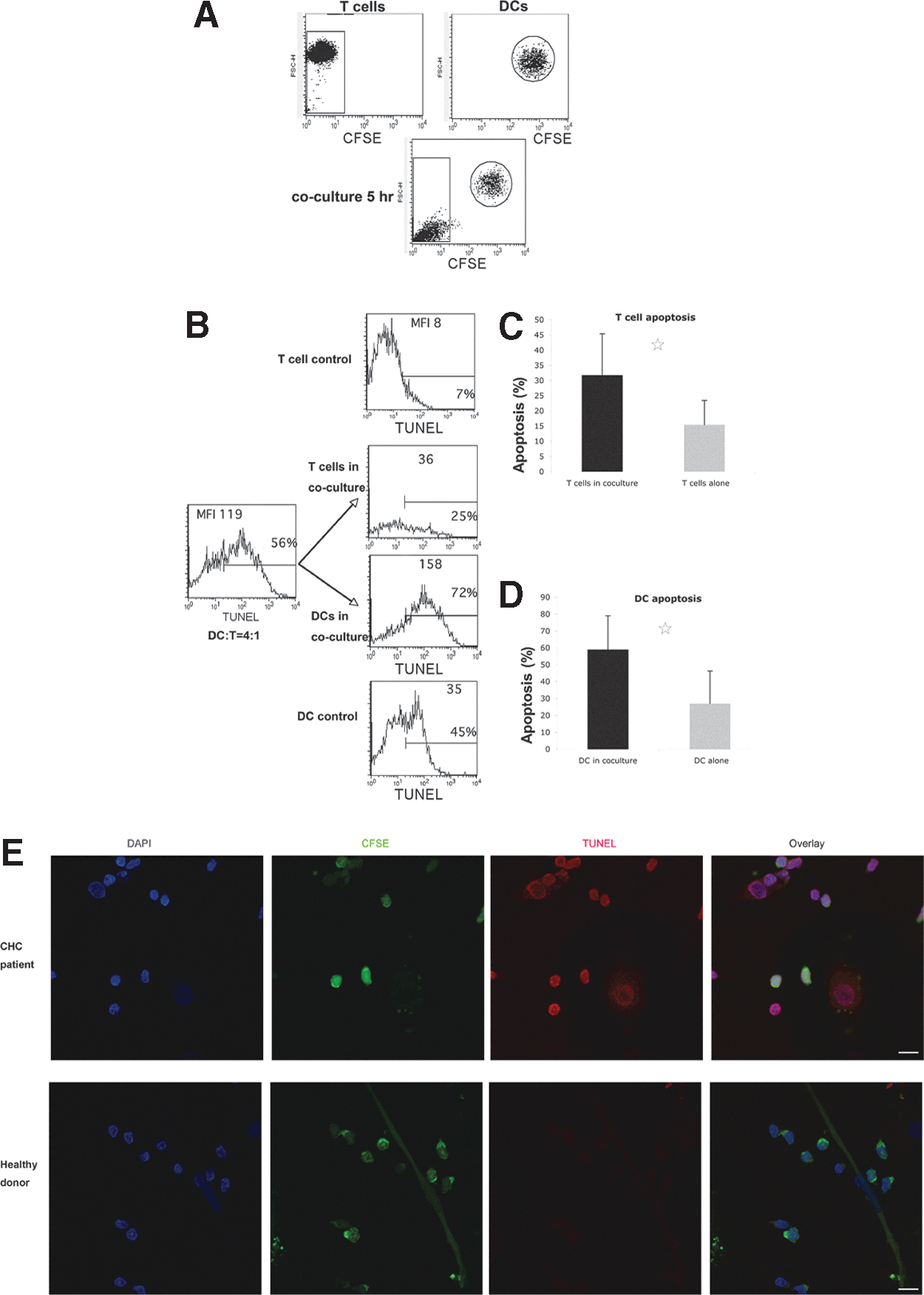
Increased apoptosis of both healthy CD4 T cells and CHC patient moDCs after co-culture.
Healthy CD4 T cells (CFSE- gating) co-cultured with CHC patient moDCs had more apoptosis than the same healthy CD4 T cells that were cultured alone, indicating CHC patient moDCs induced apoptosis of CD4 T cells (Fig. 5B, C, and E). CHC patient moDCs (CFSE+ gating) in the co-culture also had more apoptosis than the same moDCs that were not co-cultured (Fig. 5B, D, and E). This suggested that moDCs might depend on viable T cells to survive in vitro, and apoptotic T cells might induce moDCs to undergo apoptosis.
Healthy T cells and healthy moDCsin co-culture demonstrated comparable levels of apoptosis compared to these cells being cultured alone (Supplementary Fig. S1; Supplementary material is available online at
Specific antibodies and transwell membranes can block the killing of CHC patient moDCs
CHC patient moDCs demonstrated increased expression of FasL and PD-L2, and had a killing effect on peripheral blood T cells. We hypothesized that the killing effect of patient moDCs may be mediated by the interactions between certain inhibitory ligands (FasL and PD-L2) expressed on moDCs and the corresponding receptors (Fas, PD-1) expressed on target T cells, and if so, antibodies against these molecules may abolish the killing effect.
To test this hypothesis, antibodies against FasL and PD-L2 and their corresponding receptors were used to block the interaction between these molecules. It was found that antibodies against FasL, Fas, PD-L2, and PD-1 all partially abolished the killing effect, but isotype control antibodies did not (Fig. 6). These results suggested that the killing effect was mediated by the interactions between FasL/Fas and PD-L2/PD-1.
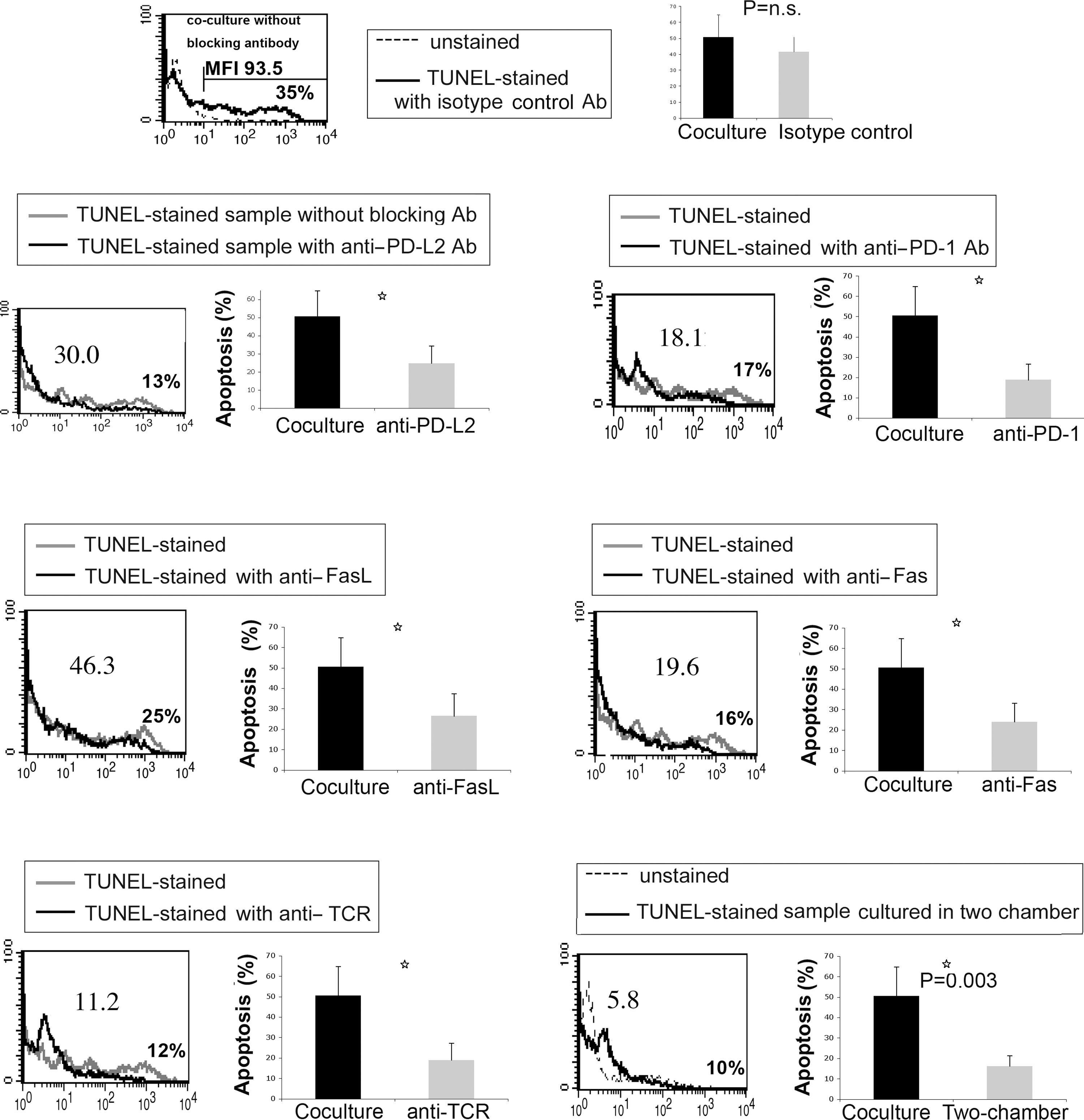
The cytotoxic activity of moDCs from CHC patients can be abolished either by a transwell membrane, or by specific antibodies against Fas/FasL, PD-1/PD-L2, and TCR. In the blocking assay, cells were pre-incubated with antibodies for 45 min before mature moDCs from CHC patients and allogeneic CD4 T cells from healthy donors were co-cultured. In the transwell assay, CHC patient moDCs and healthy CD4 T cells (2.5×105 cells/well) were cultured in two chambers isolated by a transwell permeable support (0.4 μm polycarbonate membrane). The ratio of moDC: CD4 T-cell was 4:1. The cells were co-cultured for 5 h before TUNEL staining. Numbers indicate the MFI of TUNEL-stained samples. The marker is set to make the percentage of positive cells of unstained controls (background)<1%. The percentage of positive cells of TUNEL-stained samples is shown. This flow cytometry plot is representative of five individual experiments. The statistical data was analyzed by unpaired t test (n=5) and is expressed as mean±SD (p=0.0094 for blocking PD-L2, p=0.0053 for blocking PD-1, p=0.016 for blocking FasL, p=0.0075 for blocking Fas, p=0.035 for blocking TCR, and p=0.003 for blocking with two-chambers).
Unexpectedly, the antibody against TCR also partially blocked the killing (Fig. 6). This suggested that the killing effect might require cell–cell contact, since the interaction between TCR and HLA molecules is important for contact between DCs and T cells (37).
Furthermore, the killing effect was almost totally blocked when moDCs and T cells were cultured in two chambers of transwell plates separated by polycarbonate membranes (Fig. 6), which confirmed that the killing of patient moDCs primarily required cell–cell contact rather than being mediated by soluble effector molecules (e.g., TNFα).
Effect of CHC patient moDCs on Jurkat T cells
To test if CHC patient moDCs are cytotoxic to multiple target cells, a human leukemia T cell line, Jurkat T cells, was selected as a target T cell line. Being sensitive to Fas-mediated apoptosis, Jurkat T cells have been widely used as a target in killing assays (38 –40). Surprisingly, when patient moDCs were co-cultured with Jurkat T cells, the apoptosis rates were comparable to those of healthy donor moDCs co-cultured with Jurkat T cells. The same result was observed at different mDC: T-cell ratios ranging from 8:1 to 1:4 (Fig. 7A and B). These results suggest that CHC patient moDCs may specifically kill primary T cells.
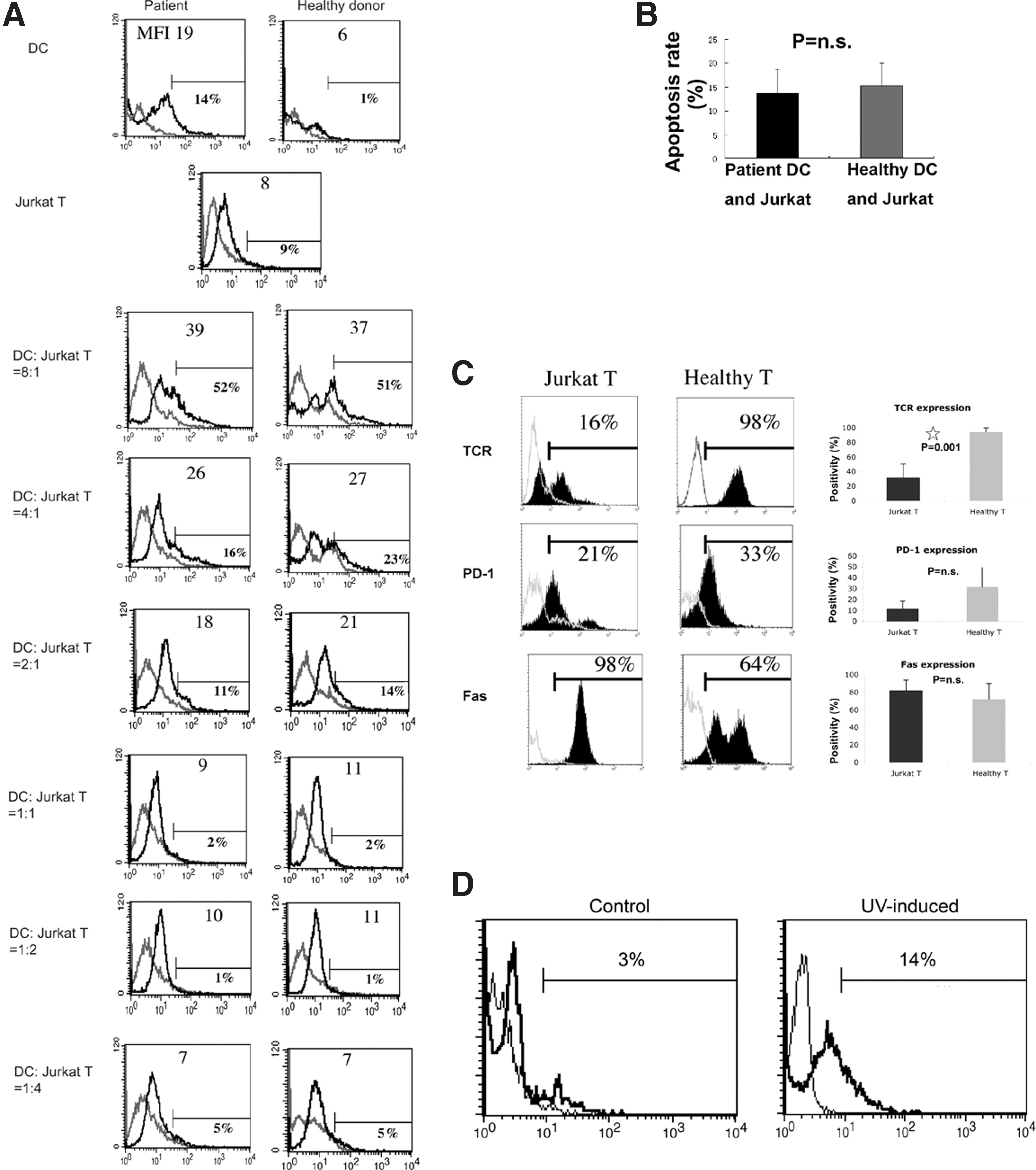
The killing effect of moDCs from CHC patients on Jurkat T cells was not seen.
As shown in Figure 6, the killing of CHC patient moDCs requires cell–cell contact, and it was primarily mediated by the interactions between FasL/Fas and PD-L2/PD-1. It was hypothesized that Jurkat T cells express one or more important cell surface molecules at different levels, which leads to their poor cell–cell contact with moDCs and their lack of sensitivity to the killing of CHC patient moDCs. To test this hypothesis, the phenotype of Jurkat T cells was examined. We found that Jurkat T cells expressed TCR at a lower level than healthy CD4 T cells (Fig. 7C).
There was the slight possibility that the Jurkat T cells used in our study were not sensitive to any apoptosis-inducing agents, or that the TUNEL method used to detect apoptosis was not appropriate for Jurkat T cells. To test these possibilities, Jurkat T cells were exposed to UV light that leads to DNA damage and thus induces cells to undergo apoptosis. We found that Jurkat T cells were sensitive to UV light-induced apoptosis (Fig. 7D). This result showed the lack of sensitivity of Jurkat T cells to the killing of patient moDCs might be due to their poor expression of TCR, which facilitates the cell–cell contact of Jurkat T cells with moDCs.
Patient moDCs have a killing effect on autologous patient T cells
In the previous assays, healthy CD4 T cells were used as the target cells. Since autologous CHC patient T cells are the real targets of the patient DCs, the cytotoxic activity of patient moDCs on autologous patient CD4 T cells was to be examined.
We first examined the spontaneous apoptosis of patient CD4 T cells. We found that CD4 T cells from CHC patients have a higher level of spontaneous apoptosis than CD4 T cells from healthy donors. Furthermore, cell apoptosis increased in the autologous coculture of patient moDCs and T cells (Supplementary Fig. S2). Next, patient moDCs were CFSE-labeled before co-culture. Patient CD4 T cells (CFSE- gating) have a significantly higher level of apoptosis in co-culture compared to the same T cells without co-culture (Fig. 8A–C). These results indicate that patient moDCs could kill autologous patient CD4 T cells.
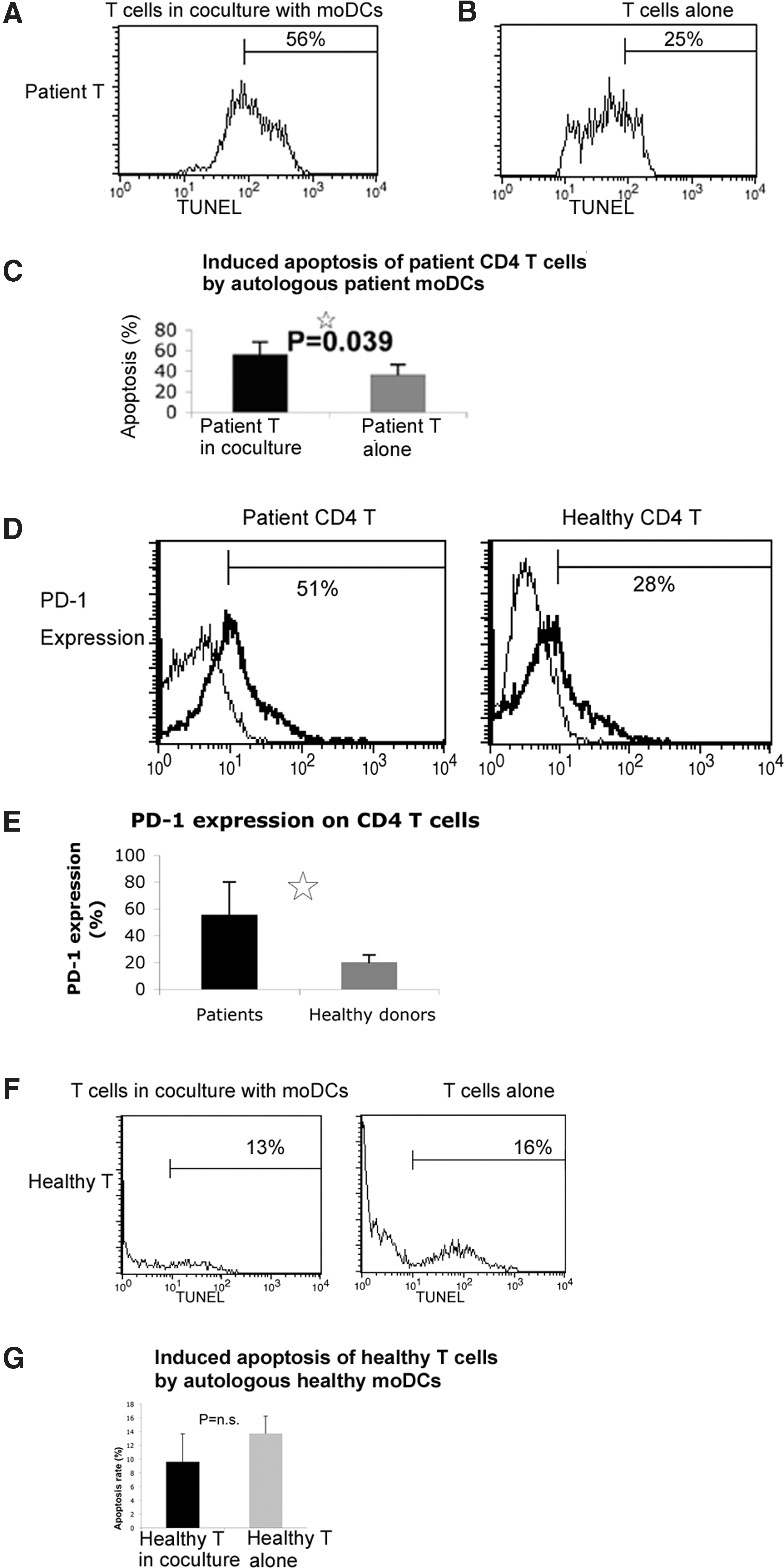
moDCs from CHC patients can induce the apoptosis of autologous patient CD4 T cells.
The phenotype of patient CD4 T cells was analyzed. CHC patient CD4 T cells demonstrate increased expression of PD-1 compared to healthy CD4 T cells (Fig. 8D and E). This indicates that patient CD4 T cells are more sensitive than healthy CD4 T cells to the cytotoxic activity of patient moDCs.
In the previous assays, co-culture of CHC patient moDCs with autologous patient CD4 T cells resulted in the increased cell apoptosis. There was a possibility that co-culture of autologous immune cells may lead to the cytotoxic effect. To test the possibility, healthy moDCs were co-cultured with autologous healthy CD4 T cells. HealthyT cells in co-culture demonstrated a comparable level of apoptosis compared to these cells being cultured alone (Fig. 8F-G). This result excluded the possibility that co-culture of healthy moDCs and CD4 T cells always leads to increased cell apoptosis. The data further support the concept that patient moDCs have upregulated cytotoxic activity compared to healthy moDCs.
Both CD4 (41)and CD8 T-cell responses (42) are important for HCV clearance. After the assays using CD4 T cells as target cells, we set out to determine whether CD8 T cells are sensitive to cytotoxic activity of moDCs from CHC patients.
CD8 T cells from CHC patients in co-culture with autologous patient moDCs had a higher level of apoptosis than the same CD8 T cells without co-culture (Supplementary Fig. S3A-C). This indicates that CD8 T cells were also sensitive to the cytotoxic activity of CHC patient moDCs.
Ex vivo mDCs from CHC patients demonstrate upregulated cytotoxic activity compared to ex vivo mDCs from healthy donors
Our previous assays have demonstrated that in vitro generated moDCs from CHC patients have a changed phenotype (increased expression of FasL and PD-L2) and increased cytotoxic activity compared to moDCs from healthy donors. However, the moDCs were monocyte-derived in vitro. Although moDCs have been widely used to represent mDCs, it is possible that moDCs are different from ex vivo isolated mDCs in certain aspects. To examine the phenotype and cytotoxic activity of mDCs circulating in peripheral blood of CHC patients, mDCs were ex vivo isolated from peripheral blood using MACS.
Ex vivo isolated mDCs from CHC patients have increased expression of FasL and PD-L2 compared to ex vivo isolated mDCs from healthy donors (Fig. 9A). To determine the cytotoxic activity of ex vivo isolated patient mDCs, the ex vivo mDCs were co-cultured with allogeneic healthy CD4 T cells. The cell apoptosis level was increased when CHC patient ex vivo mDCs were co-cultured with allogeneic healthy CD4 T cells compared to healthy ex vivo mDCs co-cultured with the same allogeneic CD4 T cells. There were more apoptotic CD4 T cells (CFSE-) and more apoptotic mDCs (CFSE+) in the patient co-culture compared to the cells in the healthy cell co-culture (Fig. 9B). The results indicated that mDCs ex vivo isolated from CHC patients had upregulated cytotoxic activity, which confirmed our previous results on in vitro generated moDCs.
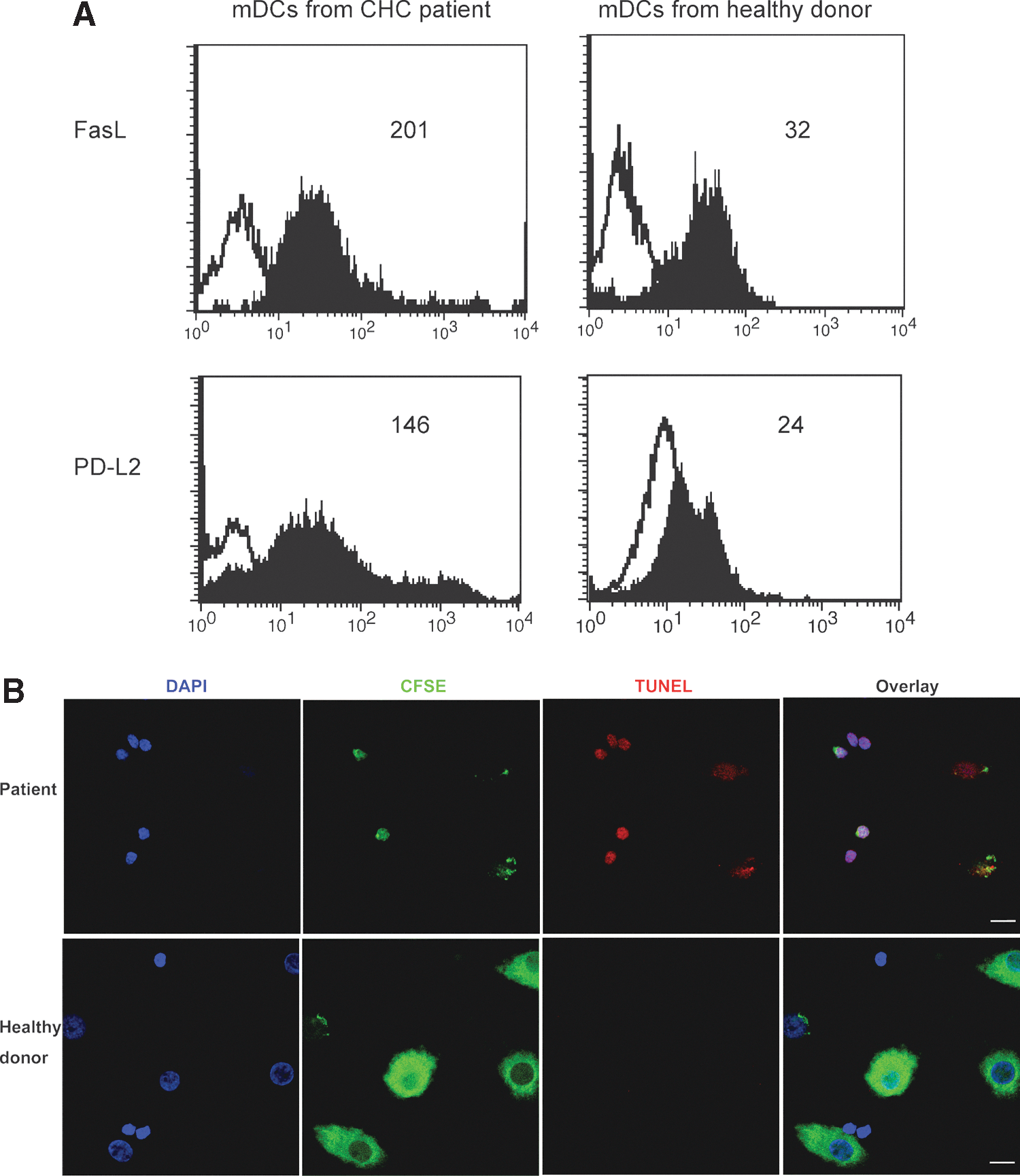
The ex vivo mDCs isolated from peripheral blood of CHC patients induce the apoptosis of healthy CD4 T cells. Ex vivo mDCs were isolated from peripheral blood using BDCA-1 positive isolation kit by MACS.
Discussion
In this study, we found the presence of killer DCs during chronic HCV infection. Previous studies have shown that DCs can induce the apoptosis of CD4 T cells during MV (17,18) and HIV (19,20) infections. MV induces TRAIL expression in DCs and these DCs induce the apoptosis of activated T cells (17) and can kill tumor cells (18). MV infection induces a profound immunosuppression that can lead to serious secondary infections. Fugier-Vivier et al. (17) observed a significant cell apoptosis in both moDCs and CD3 T cells in the co-culture of MV-infected moDCs and CD3 T cells, and suggested that DCs represent a major target of MV. These authors believed that MV suppressed cell-mediated immunity by interfering with the survival and functions of DCs and T cells. Vidalain et al. (18) demonstrated that MV infection induced TRAIL mRNA and protein expression in human moDCs, and MV-infected DCs were cytotoxic via the TRAIL pathway. However, TRAIL was not detected on the surface of DCs. Therefore, cytotoxic activity of MV-infected DCs is mostly mediated by sTRAIL. Vidalain et al. (18) also suggested that either of the two functions could be assigned to human DCs in vivo: an antigen-presenting cell function that generates effector T cells, or cytotoxic activity upon infection with immunosuppressive viruses. During HIV infection, killer DCs produce TRAIL and induce the apoptosis of CD4 T cell lines (19,20). Stary et al. (20) found the upregulated expression of membrane-bound TRAIL (mTRAIL) on pDCs during HIV infection. HIV infection was also associated with the upregulation of the apoptosis-transmitting receptor TRAIL R1 on activated CD4 T cells and therefore these T cells became susceptible to TRAIL-dependent pDC-mediated killing. Therefore, Stary et al. (20) defined pDCs as killers of CD4 T cells and believed that it implied a new mechanism of disease progression in HIV infection. In the present study, our data demonstrated that DCs had upregulated cytotoxic activity and were capable of inducing T-cell apoptosis, which supported the concept that the cytotoxic activity of DCs can be manipulated by viruses to evade host immune pressure. However, our data revealed multiple inhibitory molecules which have upregulated expression during virus infection. Different from MV and HIV infections during which TRAIL expression by DCs was upregulated, we found that the expression of membrane-bound FasL and PD-L2 on mDCs was upregulated but the expression of both mTRAIL and sTRAIL was unchanged in CHC patients.
Our study shows that mDCs from peripheral blood of CHC patients can induce the apoptosis of CD4 and CD8 T cells in vitro. We also demonstrate that CD4 T cells from CHC patients have a higher level of spontaneous apoptosis than CD4 T cells from healthy donors. This correlates with a study showing that CD8 T cells in CHC patients are highly apoptotic, which is associated with significant functional T-cell deficits (43). CD8 T cells are important effector T cells, which kill virus-infected cells and thus are crucial for virus clearance. We suppose that there are two mechanisms leading to the poor CD8 T-cell response in CHC patients: First, mDCs in CHC patients can directly kill CD8 T cells. Patient mDCs have upregulated expression of FasL and PD-L2. Patient CD8 T cells express corresponding receptors Fas and PD-1, and therefore are susceptible to the killer DCs. Second, CHC patient mDCs can affect CD4 T cells and thus weaken CD8 T-cell response indirectly. CD4 T helper cells are critical for the initiation and maintenance of CD8 T-cell response. Patient mDCs have decreased ability to stimulate CD4 T-cell proliferation (23), and patient mDCs have increased expression of inhibitory molecules (FasL and PD-L2) as well as increased cytotoxic activity that enables them to kill CD4 T cells. The poor CD4 T-cell response can also lead to poor CD8 T-cell response in CHC patients. In brief, DCs are pivotal in regulating T-cell responses, and HCV may develop multiple mechanisms to manipulate DCs and thus facilitate its evasion of host immune responses.
This study demonstrates that mDCs from CHC patients may work as killer DCs to induce CD4 and CD8 T-cell apoptosis. Although our study correlates with a previous report by Ciesek et al. that ex vivo mDCs do not increase their TRAIL production and can not kill Jurkat T cells; however, Ciesek et al. concluded that ex vivo mDCs of patients with hepatitis C displayed no cytotoxic activity (21). This discrepancy between the study by Ciesek et al. and our study may be explained by two factors: they used different target T cells, and they only examined the TRAIL molecule. First, Ciesek et al. used immortalized leukemia cell lines as target cells, including JY-EBV B cells, K562 cells, U937, and Jurkat T cells. In this study, we used primary CD4 and CD8 T cells freshly isolated from human blood as target cells. We demonstrated that the cytotoxic activity of CHC patient mDCs was upregulated, and CHC patient mDCs can induce the apoptosis of allogeneic healthy T cells and autologous CHC patient T cells. Since CD4 and CD8 T cells circulating in the blood of CHC patient are the real targets of DCs in vivo, we believe that assays using fresh T cells as targets are more representative of CHC than assays using immortalized leukemia cell lines in terms of clinical significance. Second, we studied multiple inhibitory molecules (FasL, PD-L1, PD-L2, and TRAIL) on the surface of mDCs, whereas Ciesek et al. (21) focused only on the TRAIL molecule. We found that CHC patient mDCs had upregulated expression of FasL and PD-L2, which were not examined in the study by Ciesek et al. (21).
It has been reported that PD-1 is a potential target of immunotherapy for chronic viral infections, for example, HCV (44,45) and HIV infections (46). In this study, we found that CHC patient CD4 T cells have a higher expression of PD-1 and a higher level of spontaneous apoptosis compared to healthy donor CD4 T cells. The increased expression of PD-1 on CD4 T cells from CHC patients indicates that T cells of CHC patients are more sensitive to apoptosis than healthy donor T cells. This correlates with a previous study that HCV-specific CD8 T cells have upregulated expression of PD-1 (44,45), and undergo significant apoptosis in the peripheral blood and the liver during chronic HCV infection (43). Our findings about the increased expression of PD-1 on CD4 T cells during chronic HCV infection also correlate with the current concept that PD-1 is highly expressed on exhausted T cells during chronic viral infections (22).
The two ligands for PD-1 (PD-L1 and PD-L2) differ in their expression and predominant function. PD-L1 is expressed much more broadly than PD-L2. While PD-L1 is widely expressed on multiple blood cells and a variety of nonhematopoietic cells, PD-L2 is only expressed on DCs, macrophages, and bone marrow-derived cultured mast cells (22). Blocking PD-L2 on DCs results in enhanced T-cell proliferation, while blocking PD-L1 results in similar, but more modest effects (33). In a previous study on HCV, blocking PD-L1 restored T-cell function in HCV-infected patients (36). However, our study suggests that PD-L2 and FasL may be more important than PD-L1 in regulating CD4 and CD8 T-cell apoptosis during HCV infection. Our results correlate with the previous studies that PD-L1 is more associated with self-antigen (and thus regulates auto-immunity) while PD-L2 is more associated with environmental-antigen (and thus determines the outcome of pathogen infection) (22,47).
The BLyS molecule regulates both B-cell response (24 –26) and T-cell response (27,28). BLyS is predominantly produced by myeloid cells, including monocytes, macrophages, DCs, and neutrophils (48,49). Full-length BLyS (285 amino acids) is expressed on the plasma membrane of immune cells as mBLyS. After cleavage by polyprotein convertases, the extracellular C-terminal fragment (containing amino acids 134–285) is released as soluble BLyS (sBLyS). BLyS is critical for B cell maturation, function, and survival (24 –26). BLyS-knockout mice have significantly reduced spleen weight, markedly reduced numbers of peripheral blood B cells, and a profound reduction in total serum immunoglobulin (26). The level of sBLyS in the serum of HCV-infected patients was significantly higher than the level in healthy controls (50), and serum sBLyS level predicts the outcome of acute HCV infection (51). sBLyS level is significantly increased in acute HCV-infected patients evolving to chronic infection than in those with a self-limited course, and thus a higher sBLyS level is associated with the persistence of HCV infection (51). Recently, it has been reported that mBLyS expressed on DCs regulates T-cell responses (27,28). mBLyS expressed on APCs provides a complete and potent second signal for T cell activation and proliferation (52,53). Our previous study had shown that CHC patient mDCs have decreased expression of HLA-DR and CD86, and their ability to stimulate CD4 T-cell proliferation was impaired (23). In this study, we found that mBLyS expression on DCs was not changed during CHC infection, which indicates that mBLyS on DCs may not be one of the molecules leading to the impaired T-cell response during CHC. However, mBLyS expression on other immune cells is yet to be determined in future studies.
DCs can have an immunosuppressive effect and induce immune tolerance, which may be used by viruses to evade host immune response and thus facilitates virus persistence. During HCV infection, DCs produce IL-10 (54) and induce the proliferation of regulatory T cells (Treg) (55). In this study, we further described the cytotoxic activity of CHC patient mDCs on T cells. These studies support the observation that HCV is very successful in evading host immune response to promote chronic infection, and DCs may be pivotal in determining the outcome of HCV infection.
In conclusion, mDCs from chronic HCV-infected patients express increased levels of inhibitory ligands, FasL and PD-L2, and can induce the apoptosis of CD4 and CD8 T cells. This study demonstrates the presence of killer DCs during HCV infection, which represents a novel mechanism of HCV-induced immune dysfunction. Our study demonstrates that the function of mDCs may be switched from immunogenic to tolerogenic during chronic HCV infection. Such a switch in DC function contributes to the impaired T-cell responses in CHC patients. The information may help to design future studies to recover mDC function, which may rescue host immune responses to clear HCV infection.
Footnotes
Acknowledgments
This work was supported by grants from the Canadian Institutes of Health Research. LZ was supported by a scholarship from the National Canadian Research Training program in Hepatitis C (NCRTP-HepC). DLJT holds the CIHR/GSK Chair in Virology.
The authors thank Dr. William R. Addison for technical help. We are grateful to Craig R. Madill, Justin Shields, and Patrick V. Sullivan for proof-reading assistance.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.