
Abstract
Theiler's murine encephalomyelitis virus (TMEV) induces a demyelinating disease in susceptible SJL mice that has similarities to multiple sclerosis in humans. TMEV infection of susceptible mice leads to a persistent virus infection of the central nervous system (CNS), which promotes the development of demyelinating disease associated with an inflammatory immune response in the CNS. TMEV infection of resistant C57BL6 mice results in viral clearance without development of demyelinating disease. Interestingly, TMEV infection of resistant mice deficient in IFNγ leads to a persistent virus infection in the CNS and development of demyelinating disease. We have previously shown that the innate immune response affects development of TMEV- induced demyelinating disease, thus we wanted to determine the role of IFNγ during the innate immune response. TMEV-infected IFNγ-deficient mice had an altered innate immune response, including reduced expression of innate immune cytokines, especially type I interferons. Administration of type I interferons, IFNα and IFNß, to TMEV- infected IFNγ-deficient mice during the innate immune response restored the expression of innate immune cytokines. Most importantly, administration of type I interferons to IFNγ-deficient mice during the innate immune response decreased the virus load in the CNS and decreased development of demyelinating disease. Microglia are the CNS resident immune cells that express innate immune receptors. In TMEV- infected IFNγ-deficient mice, microglia had reduced expression of innate immune cytokines, and administration of type I interferons to these mice restored the innate immune response by microglia. In the absence of IFNγ, microglia from TMEV-infected mice had reduced expression of some innate immune receptors and signaling molecules, especially IRF1. These results suggest that IFNγ plays an important role in the innate immune response to TMEV by enhancing the expression of innate immune cytokines, especially type I interferons, which directly affects the development of demyelinating disease.
Introduction
Theiler's murine encephalomyelitis virus (TMEV) is a natural mouse pathogen that can trigger the development of demyelinating disease in susceptible mouse strains and has immunological and pathological similarities to MS. Infection of susceptible SJL mice with the BeAn strain of TMEV leads to a persistent virus infection of the CNS, predominantly in the microglia/macrophage population (22,32). The persistent virus infection of the CNS leads to the development of a chronic progressive demyelinating disease with clinical signs of disease beginning around 35 days post infection. TMEV- induced demyelinating disease has been shown to be associated with a myelin-specific CD4+ T cell response directed against immunodominant myelin antigen, PLP139–151, which can be detected around 50–55 days post infection (14). As the demyelinating disease progresses, epitope spreading continues to initiate CD4+ T cell responses against additional myelin antigens, including PLP56–70, MOG92–106, PLP178–191, and MBP84–104 (14,26). We have shown that the innate immune response to TMEV has a direct effect on development and progression of demyelinating disease (34). TMEV infection of resistant mouse strains, such as C57BL6 mice, does not result in demyelinating disease due to efficient clearance of the virus from the CNS (29).
The innate immune response is initiated through cellular receptors, such as Toll like receptors (TLR), which recognize pathogen-associated molecular patterns. The innate immune response initiates the expression of cytokines and chemokines that activate and recruit immune cells to the site of infection. Thus, the innate immune response directly affects the development of the adaptive immune response. Type I interferons, IFNα and IFNß, are the predominant cytokines expressed after virus infections. Type I interferons have direct antiviral actions that disrupt viral replication, however, type I interferons also have immunomodulatory functions. IFNα and IFNß promote NK cell-mediated cytotoxicity, as well as blastogenesis and proliferation (6). IFNα and IFNß drive the maturation of dendritic cells that promote development of Th1 type CD4+ T cells secreting IFNγ and promote proliferation of CD4+ T cells (5,39). Additional, studies have shown that type I interferons promote CD8+ T cell proliferation and survival as well as enhance the B cell response (19,40). Most interesting, IFNß is used as a treatment for MS, however, the mechanism by which IFNß modulates disease has not been completely determined. We have previously shown that IFNß expression during the innate immune response to TMEV decreased development and progression of demyelinating disease (34).
IFNγ is a type II interferon that has immunomodulatory functions and is produced by several types of cells, including T cells and natural killer cells. IFNγ expressed during the immune response activates macrophage to increase expression of pro-inflammatory cytokines and to upregulate expression of MHC class II and co-stimulatory molecules for antigen presentation. IFNγ is most often associated with Th1 type CD4+ T cell response and is involved in development of Th1 versus Th2 type responses (13,38). CD8+ T cells also secrete IFNγ, which has been suggested to mediate the noncytolytic functions of these cells, especially in clearance of CNS virus infections (24,36). Most recently, IFNγ has been shown to have antiviral properties by promoting expression of antiviral factors through activation of IFN-stimulated genes (ISGs) (9,23). Further, IFNγ has been suggested to play a role in the innate immune response through TLR-dependent IFNγ signaling pathway during virus infections (30,31). IFNγ has been shown to induce the expression of transcription factor, interferon regulatory factor 1 (IRF-1), which can activate the expression of type I interferons, especially IFNß, and cytokines, such as IL-12 (15,28).
The role for IFNγ during demyelinating disease has been examined using mouse models of MS. In the experimental autoimmune encephalomyelitis (EAE) model, the loss of IFNγ or IFNγ receptor in mice results in development of EAE in resistant mouse strains or worsening of disease in susceptible mouse strains (11,16,20). Similarly in the TMEV model, studies with the DA strain of TMEV have demonstrated that mice resistant to TMEV-induced demyelinating disease develop demyelination in the absence of IFNγ receptor or IFNγ expression (12,37). Based on recent reports of antiviral functions of IFNγ, we wanted to examine how IFNγ affects the innate immune response and development of demyelinating disease in IFNγ-deficient mice infected with the BeAn strain of TMEV. In these studies, we show that IFNγ-deficient mice, on the resistant strain (C57BL6), infected with TMEV develop demyelinating disease similar to the susceptible mice (SJL). Most interestingly, TMEV-infected IFNγ-deficient mice have a reduced innate immune response marked by reduced expression of innate immune cytokines, most significantly type I interferons. Administration of type I interferons to IFNγ-deficient mice during the innate immune response to TMEV restores the innate immune response and reduces the development and progression of demyelinating disease in the IFNγ- deficient mice. Since microglia are the CNS resident innate immune cells, we showed that the innate immune response by microglia was reduced in IFNγ- deficient mice and was associated with reduced innate immune receptor and signaling molecule expression, especially IRF1. Thus, IFNγ plays an important role in the innate immune response by influencing the expression of innate immune cytokines and chemokines, which directly affects the development and progression of demyelinating disease.
Materials and Methods
Infection of mice
IFNγ-deficient mice on the C57BL6 background and C57BL6 mice were purchased from Jackson Laboratories. The mice were housed at University of Wisconsin Research Animal Resource Center according to university and ACUC approved protocols. Female mice, 5–6-weeks old, were infected by intracerebral injection with 2×106 PFU of BeAn strain of TMEV. On the day of infection and 4 days post infection, mice were administered by intraperitonial (i.p.) injection of IFNα (5000 U) and IFNß (5000 U) (R&D Systems) as previously determined (34). Mice were followed for clinical disease signs, and scores were assigned based on a scale of 0–5: score 1, mice show mild waddling gait; score 2, mice show more severe waddling gate; score 3, mice had a loss of righting ability associated with spastic hind limbs; score 4, mice had paralysis of hind limbs associated with dehydration; score 5- mice were moribund.
Viral plaque assay
The brain and spinal cord were removed from the infected mice at the indicated days post infection. The organs were homogenized and diluted in serum-free DMEM. BHK-21 cells were infected with the homogenized tissue dilutions and overlaid with a 2% agar/DMEM solution and incubated at 34°C for 5 days. The cells were fixed with methanol and stained with crystal violet solution (0.12% crystal violet). The plaques were counted on each plate and multiplied by the dilution and the amount of homogenate added to the plate to determine the PFU/ml. The weight of the tissue (mg) and homogenate volume was then used to calculate PFU/mg.
RNA isolation and real time PCR
Brain and spinal cord were removed from mice at the indicated days post infection. The RNA was isolated from the organs using Trizol (Invitrogen) and was DNAse treated (Invitrogen). The RNA was converted into cDNA using oligo(dT)12–18 primers with the Transcriptor First Strand cDNA Synthesis Kit (Roche Applied Science). Real time PCR reactions were conducted with FastStart SYBR Green Master Mix (Roche Applied Science). Briefly, 0.5 μM primers, 1X SYBR Green Master Mix, and 2 μL diluted cDNA were combined, and reactions were conducted in triplicates. The primers sequences have been previously published (34). Real time PCR was conducted on a Rotorgene 6000 (Corbett Research) using a hot start with cycle conditions, 40 cycles; 95°C 15 sec, 60°C 10 sec, and 72°C 15 sec; followed by a melt from 75°C to 95°C. Quantitation of the mRNA was based on standard curves derived from cDNA standards for each primer set from 1 fg/μg of RNA to 100 ng/μg of RNA. Positive and negative cDNA controls were used for each primer set derived from known cell sources for each cytokine.
Flow cytometry and sorting
Mice underwent cardiac perfusion with PBS at the indicated days post infection, and the brain and spinal cord were removed. The brain and spinal cord were minced and digested with collagenase type IV (Invitrogen) and DNAse (Invitrogen). The organs were further dissociated and separated on a 70/30 Percoll gradient. The mononuclear cells were washed with FACS buffer (PBS with 5% normal goat serum) and blocked with antibody to CD16/32 (BD Bioscience). The cells were then incubated with fluorescently labeled antibodies specific for CD4, CD8, CD11b, CD11c, CD45, B220, Gr-1, or NK1.1. The cells were analyzed on a FACScalibur (BD Bioscience). The CD45 positive cells were analyzed to determine the number of CD4+ T cells, CD8+ T cells, macrophages, dendritic cells, B cells, NK cells, and CD11b+Gr-1+ cells in the CNS. Microglia were sorted by incubating the mononuclear cells with antibodies for CD45 and CD11b and then sorted using FascAria based on expression of CD45 intermediate and CD11b+ cells.
T cell assays
Spleens were removed from infected mice and dissociated through a stainless steel screen to obtain a homogenous cell suspension, and the red blood cells were lysed. CNS cells were isolated from the brain and spinal cord as described above. ELISPOT assays were performed with antibody for IL-2 (BD Bioscience). The cells were cultured at 2.5 x 105 cells per well (96-well plates) in HL-1 medium (BioWhittaker) supplemented with 2 mM L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin. Peptides were added to the wells at increasing concentrations from 1-100 μM VP421–40 (INNFYSNQYQNSIDLSASGG) (CPC Scientific). The amino acid composition was verified by mass spectrometry, and purity was assessed by HPLC
Statistical analysis
A statistical comparison of the percentage of animals showing clinical disease between any two groups of mice will be performed by χ2 test using Fisher's exact probability. Comparison between groups in the immunological assays was determined using unpaired Student's t test (p<0.05).
Results
IFNγ-deficient mice on the resistant mouse strain develop TMEV-induced demyelinating disease
SJL mice infected with the BeAn strain of TMEV develop an inflammatory demyelinating disease, which is used as an animal model for MS. On the contrary, C57BL6 mice infected with the BeAn strain of TMEV do not develop demyelinating disease. To determine the role of IFNγ in the innate immune response during TMEV- induced demyelinating disease, C57BL6 mice deficient in IFNγ were infected with TMEV (8 mice per group) (Fig. 1A). The TMEV-infected IFNγ- deficient mice developed clinical signs of demyelinating disease with similar characteristics as the susceptible SJL mice, including loss of righting ability and ascending spastic paralysis (34). SJL mice infected with TMEV develop a persistent virus infection in the CNS associated with development of demyelinating disease. On the contrary, C57BL6 mice infected with TMEV clear the virus from the CNS early during infection. Thus, IFNγ-deficient mice (3 mice per group) were examined for virus load in the brain and spinal cord at various times following infection (Fig. 1B and 1C). Interestingly, TMEV- infected IFNγ-deficient mice were unable to clear the virus from the brain and spinal cord, resulting in a persistent virus infection of the CNS. The virus load in the brain and spinal cord of TMEV-infected IFNγ-deficient mice during demyelinating disease was similar to levels observed in TMEV-infected SJL mice (7). Thus, IFNγ plays an important role in the resistance of C57BL6 mice to TMEV-induced demyelinating disease, since IFNγ-deficient mice on the C57BL6 background develop demyelinating disease associated with a persistent virus infection of the CNS.
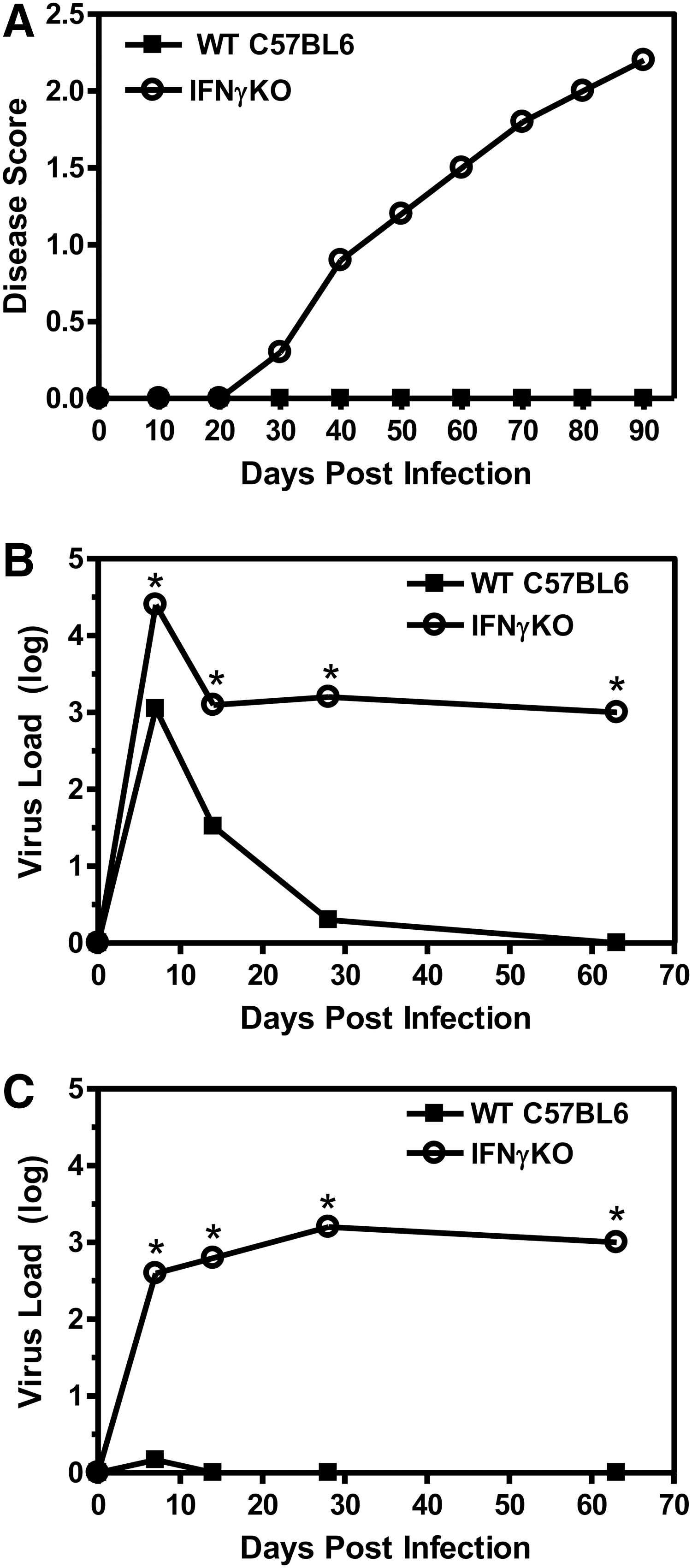
Resistant C57BL6 mice deficient in IFNγ develop TMEV-induced demyelinating disease. C57BL6 wild-type or IFNγ-deficient mice (10 mice per group) were infected with BeAn strain of TMEV. Infected mice were examined for clinical signs of demyelinating disease and scored on a disease scale of 0–5
IFNγ-deficient mice have a reduced innate immune response in the CNS after TMEV infection
Since the IFNγ-deficient mice on the C57BL6 background develop a persistent virus infection, we wanted to determine whether the innate immune response to virus infection was altered in mice deficient in IFNγ compared to wild-type mice. The innate immune response to virus infections induces the expression of type I interferons, IFNα and IFNß, as well as other innate immune cytokines. Thus, the expression of cytokines, IFNα, IFNß, IFNγ, TNFα, IL-12, and IL-6, was examined in the CNS during the innate immune response and the early adaptive immune response following TMEV infection of wild-type and IFNγ-deficient mice (3 mice per group) (Fig. 2). TMEV-infected wild-type C57BL6 mice expressed IFNγ in the CNS immediately after infection during the innate immune response and continued to increase expression of IFNγ until peaking during the adaptive immune response to virus infection at 7 days post infection. TMEV-infected C57BL6 mice expressed high levels of IFNα and IFNß, as well as innate immune cytokines, TNFα, IL-6, and IL-12, immediately following TMEV infection. Most interestingly, TMEV-infected IFNγ-deficient mice had dramatically reduced expression of IFNα and IFNß during the innate immune response compared to wild-type mice. Similarly, TMEV-infected IFNγ-deficient mice had reduced expression of TNFα, IL-6, and IL-12 during the innate immune response and early adaptive immune response compared to wild-type mice. These results show that IFNγ-deficient mice have reduced innate immune cytokine expression in the CNS, especially type I interferons, during TMEV infection compared to wild-type mice.
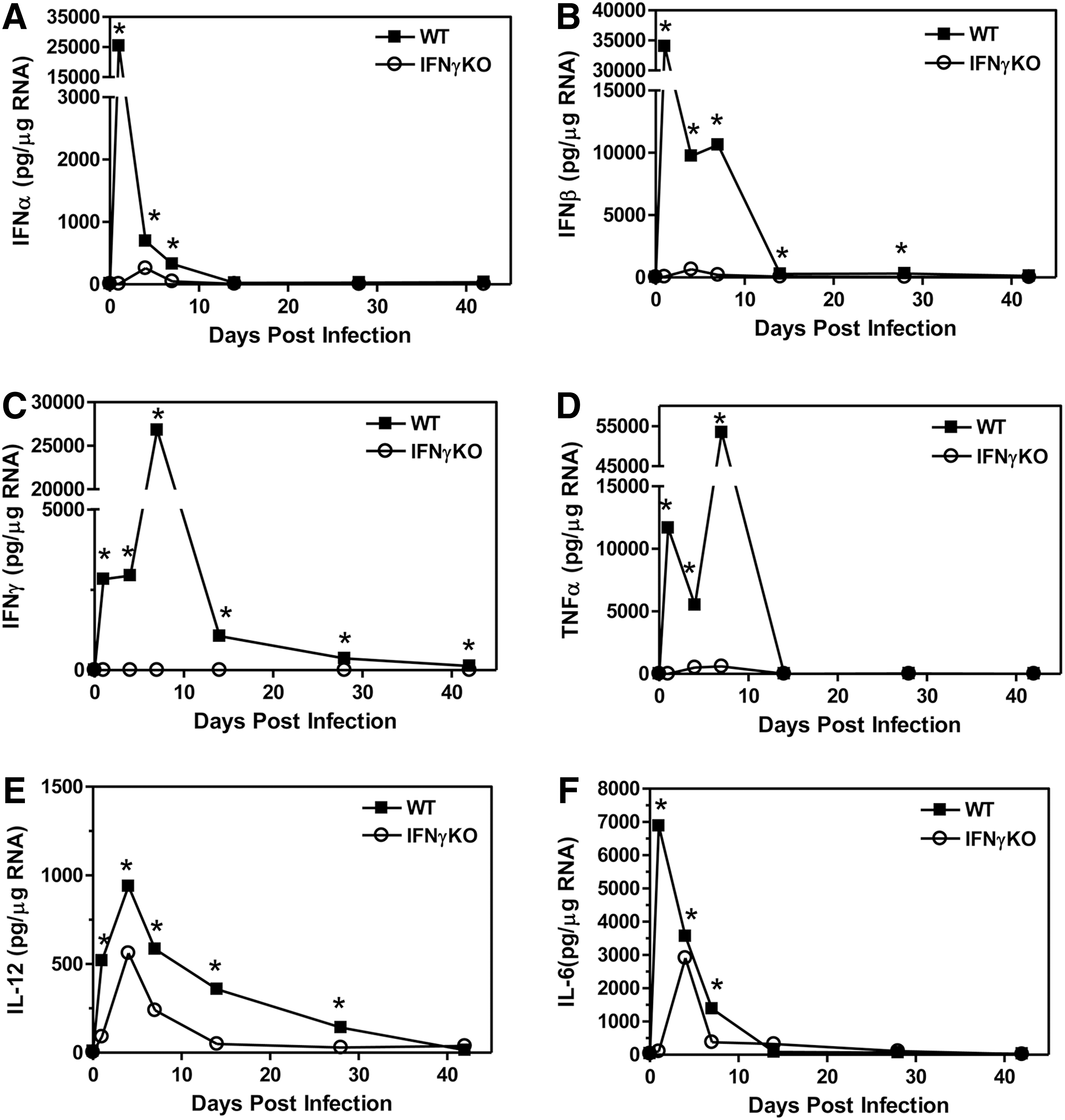
TMEV-infected IFNγ-deficient mice have reduced expression of cytokines into the CNS. C57BL6 wild-type or IFNγ-deficient mice were infected with TMEV. The brain was removed from mice (3 mice per group) at 1, 4, 7, 14, 28, or 42 days post infection. The RNA was isolated, DNAse treated, and 1 μg of RNA was converted to cDNA, and real time PCR was conducted using primers for IFNα
The innate immune response to infection is initiated by resident cells in the CNS, such as the microglia, however, peripheral immune cells immediately infiltrate into the CNS to participate in the innate immune response and continue to infiltrate into the CNS during the adaptive immune response. We wanted to determine whether the number and type of immune cells infiltrating into the CNS after TMEV infection were altered in the IFNγ-deficient mice compared to wild-type mice. The brain and spinal cord were removed from TMEV-infected wild-type and IFNγ-deficient mice (3 mice per group) to determine the infiltrating immune cells during the innate immune response and early adaptive immune response (Fig. 3). TMEV-infected IFNγ-deficient mice had decreased numbers of infiltrating macrophage in the CNS during the innate immune response but had similar numbers of macrophage during the adaptive immune response compared to wild-type mice (Fig. 3A). Gr-1 is an antibody that recognizes Ly6C and Ly6G, which are surface markers expressed on immature myeloid cells and neutrophils. TMEV-infected IFNγ-deficient mice had decreased numbers of CD11b+Gr-1+ cells in the CNS during the innate immune response compared to wild-type mice (Fig. 3B). The infiltration of dendritic cells was slightly increased, although not significantly, in the TMEV-infected IFNγ-deficient mice during the innate immune response and the adaptive immune response (Fig. 3C). TMEV-infected IFNγ–deficient mice had slightly increased but not significantly different numbers of CD8+ T cells in the CNS at 7 days post infection compared to wild-type mice, however they had similar numbers of CD8+ T cells in the CNS as wild-type mice by 14 days post infection (Fig. 3E). Finally, TMEV-infected IFNγ-deficient mice had similar numbers of CD4+ T cells and B cells infiltrating into the CNS as wild-type mice (Fig. 3F and 3G). Overall, these results show that TMEV-infected IFNγ-deficient mice have reduced innate immune cell infiltration into the CNS compared to wild-type mice, specifically the CD11b+ cells, macrophages, and Gr-1+ cells.
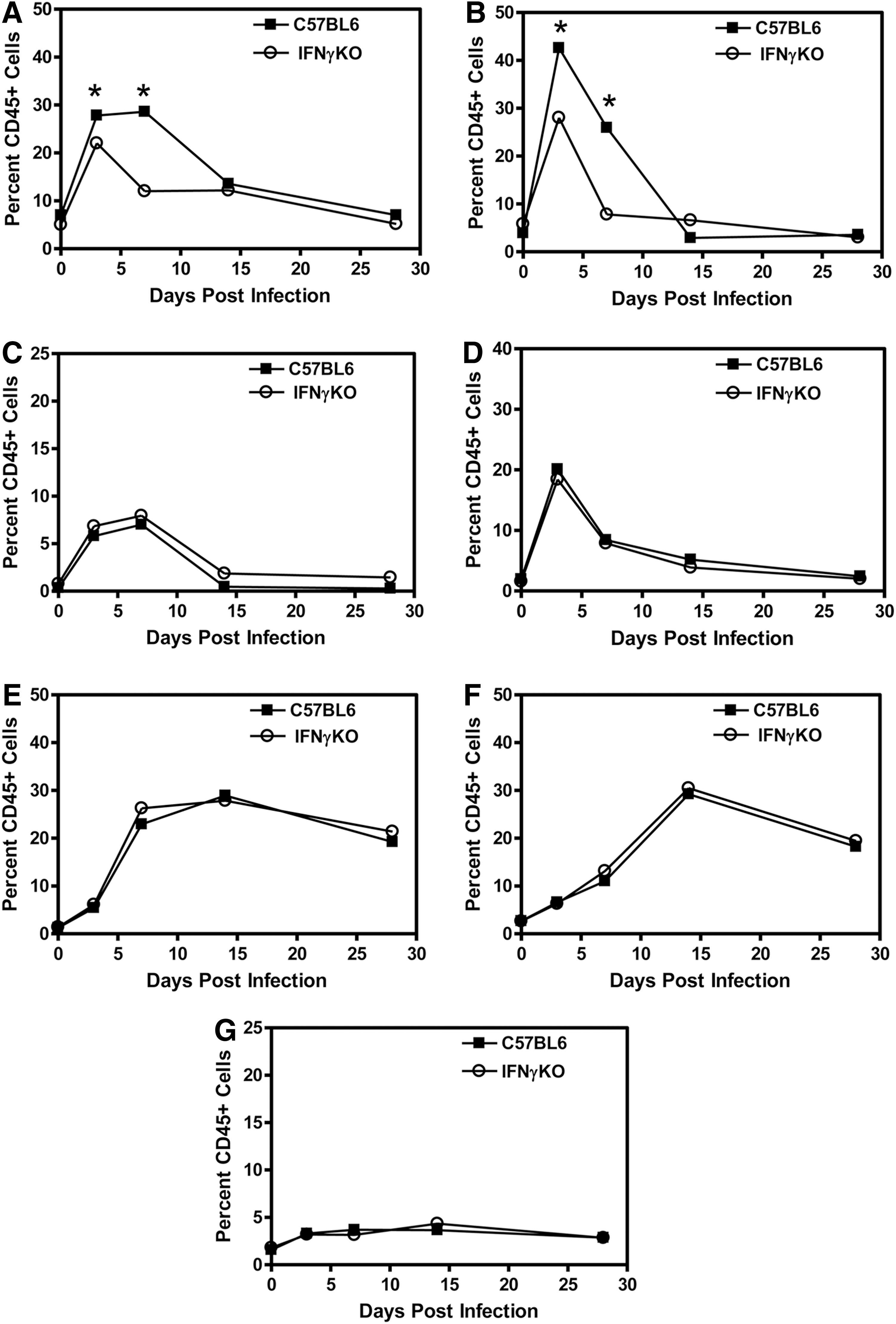
TMEV- infected IFNγ-deficient mice have reduced innate immune cell infiltration into the CNS. C57BL6 wild-type or IFNγ-deficient mice were infected with TMEV. The brain and spinal cord were removed from mice (3 mice per group) at 3, 7, 14, or 28 days post infection. The mononuclear cells were isolated and stained with fluorescently labeled antibodies for CD45, CD11b, Gr-1, CD11c, NK1.1, CD4, CD8, or B220. The cells were analyzed by flow cytometry and gated on CD45 high expressing cells. The percent of macrophage (CD11b+Gr-1- cells)
TMEV-infected IFNγ-deficient mice develop T cell responses against TMEV
IFNγ has been shown to play an important role in the Th1 type CD4+ T cell response as well as the CD8+ T cell response. TMEV infection of mice leads to the development of a Th1 type CD4+ T cell response and a CD8+ T cell response against viral antigens (25). Therefore, we wanted to determine whether mice deficient in IFNγ had a defect in the CD4+ T cell response against TMEV. The spleens, brains, and spinal cords were removed from the infected mice at 7 days post infection (3 mice per group) to examine the CD4+ T cell response against TMEV, using an IL-2 Elispot assay with virus-specific epitope VP421–40 (Fig. 4A and 4B). TMEV-infected IFNγ-deficient mice had similar virus-specific CD4+ T cell responses compared to wild-type mice in both the spleen and CNS. Next, we wanted to determine whether mice deficient in IFNγ had a defect in the CD8+ T cell response. Wild-type and IFNγ-deficient mice were infected with TMEV and an in vivo lysis assay was conducted at 7 days post infection (Fig. 4C). The mice were administered splenocytes coupled to TMEV-specific epitope, VP2121–130, or nonspecific epitope, VP270–86, and the mice were examined the next day to determine the number of virus-specific target cells that were lysed. Interestingly, TMEV-infected IFNγ-deficient mice were able to lyse the virus-specific target cells similar to wild-type mice, suggesting a similar virus-specific CD8+ T cell response. Furthermore, antigen presenting cells in the CNS were determined to be similarly activated in the wild-type and IFNγ-deficient mice following TMEV infection (data not shown). These results suggest that the lack of IFNγ did not affect the ability of mice to activate CD4+ T cell and CD8+ T cell responses against TMEV.
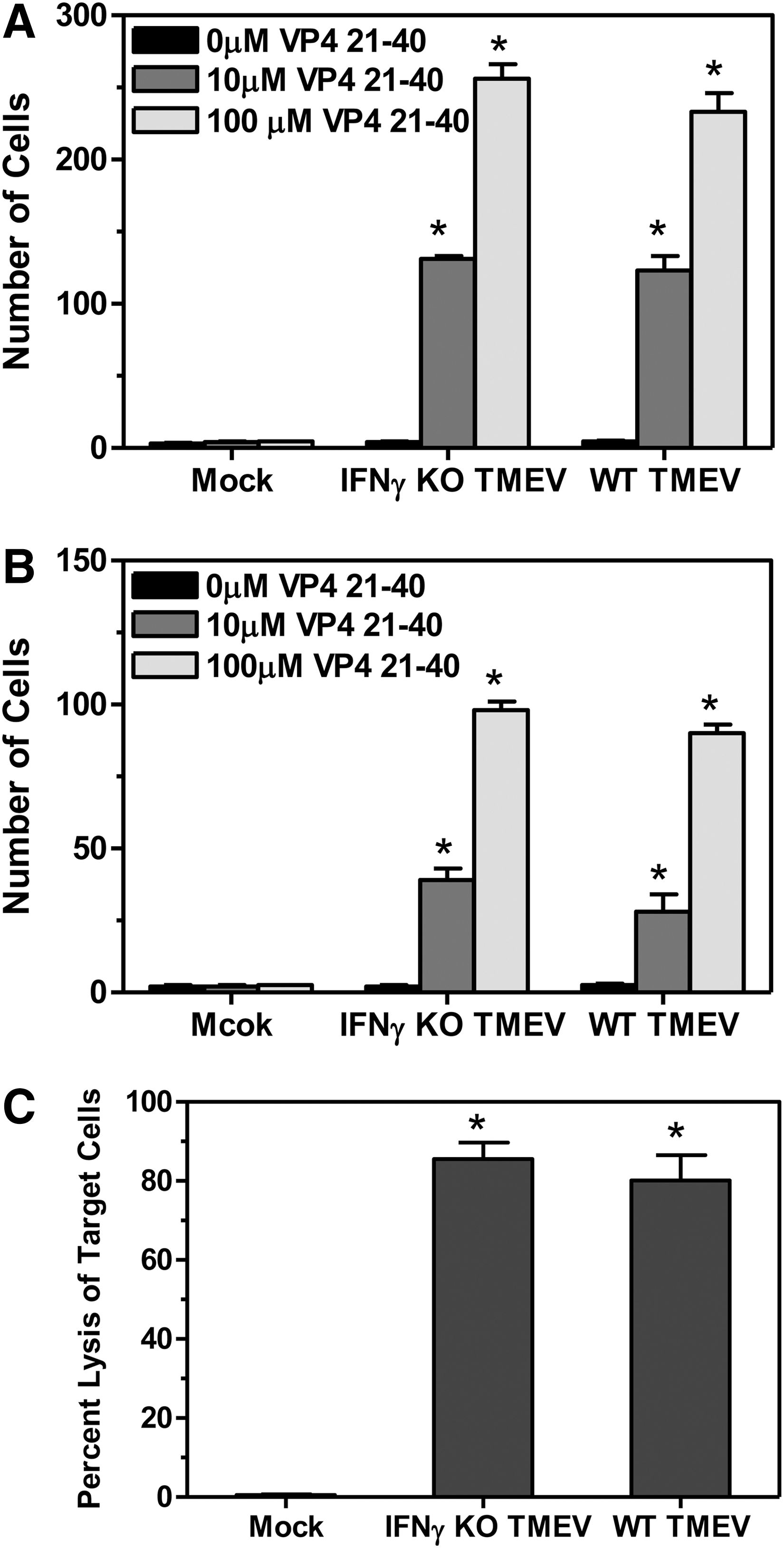
Virus-specific T cell responses following TMEV infection of IFNγ-deficient mice. Wild-type or IFNγ-deficient mice were infected with TMEV. The spleen, brain, and spinal cord were removed from mice (3 mice per group) on 7 days post infection. The mononuclear cells were isolated from the brain and spinal cord. The spleen cells
IFNγ-deficient mice develop TMEV-induced demyelinating disease associated with an inflammatory immune response in the CNS
Our results have shown that TMEV-infected IFNγ-deficient mice develop clinical demyelinating disease. Previous studies have shown that IFNγ-deficient mice develop demyelination in the spinal cord following TMEV infection (12,37). TMEV-induced demyelinating disease in susceptible SJL mice is associated with an inflammatory immune response in the CNS and an autoimmune Th1 type CD4+ T cell response (14). TMEV-induced demyelinating disease in susceptible SJL mice is associated with immune cells infiltrating into the CNS which includes macrophage, T cells, and B cells. Macrophage and CD4+ T cells have been suggested to mediate demyelination in susceptible SJL mice (26). Next, we wanted to determine whether peripheral immune cells infiltrate into the CNS during TMEV-induced demyelinating disease in IFNγ-deficient mice. Wild-type and IFNγ-deficient mice (3 mice per group) were infected with TMEV, and the CNS examined for immune cell infiltration at 70 days post infection during demyelinating disease (Fig. 5). TMEV-infected IFNγ-deficient mice had large numbers of infiltrating macrophage in the CNS as well as dendritic cells compared to wild-type mice (Fig. 5A). TMEV-infected IFNγ-deficient mice also had increased numbers of B cells and CD11b+Gr-1+ cells (Fig. 5B). Most significantly, TMEV-infected IFNγ-deficient mice had increased numbers of CD4+ T cells and CD8+ T cells compared to wild-type mice (Fig. 5C). A virus-specific CD4+ T cell response could be detected throughout demyelinating disease similar to susceptible SJL mice, however, a slight but not significant response was detected against myelin antigen, MOG35–55 (data not shown). Further, TMEV-infected IFNγ-deficient mice also expressed pro-inflammatory cytokines, such as TNFα and IL-12, in the CNS during demyelinating disease (data not shown). These results show that IFNγ-deficient mice develop TMEV-induced demyelinating disease associated with an inflammatory immune response in the CNS similar to susceptible SJL mice. These results suggest that activated CD4+ T cells infiltrate into the CNS during TMEV-induced demyelinating disease and may play a role in the pathogenesis of disease.
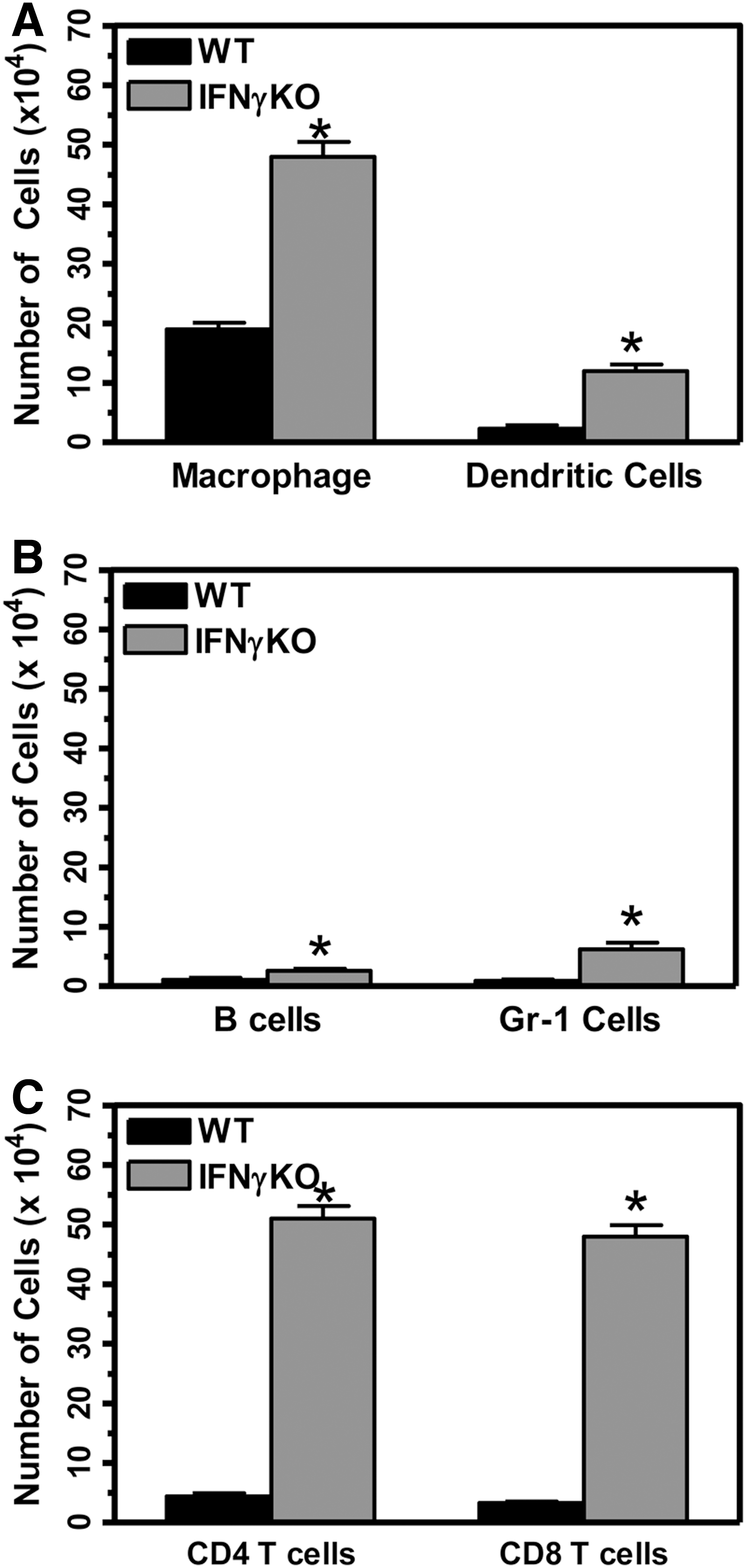
Immune responses during progressive TMEV- induced demyelinating disease in IFNγ-deficient mice. Wild-type or IFNγ-deficient mice were infected with TMEV. The brain and spinal cord were removed from the mice (3 mice per group) at 70 days post infection, and the mononuclear cells were isolated. The mononuclear cells were incubated with fluorescently labeled antibodies for CD45 and CD11b, Gr-1, CD11c, B220, CD4, and CD8. The cells were examined by flow cytometry to determine the number of macrophage (CD11b+CD11c- cells)
Administration of type I interferons to TMEV- infected IFNγ-deficient mice increases the innate immune response following TMEV infection and reduces development and progression of demyelinating disease
Our results have shown that TMEV-infected IFNγ-deficient mice develop a persistent infection in the CNS leading to development of demyelinating disease. Interestingly, we have shown that IFNγ-deficient mice have a dramatically reduced innate immune response to TMEV compared to wild-type mice. Therefore, we wanted to determine whether boosting the innate immune response in IFNγ-deficient mice after TMEV infection would alter development of demyelinating disease. Type I interferons are the predominant cytokines expressed after TMEV infection of wild-type mice, but were greatly reduced after TMEV infection of IFNγ-deficient mice. Therefore, IFNγ-deficient mice were infected with TMEV and administered type I interferons, IFNα and IFNß, during the innate immune response, on the day of infection and at 4 days post infection (8 mice per group). Most interesting, TMEV-infected IFNγ-deficient mice administered IFNα and IFNß during the innate immune response had reduced development and progression of demyelinating disease compared to control treated mice (Fig. 6A and Supplementary Table S1; supplementary material are available online at
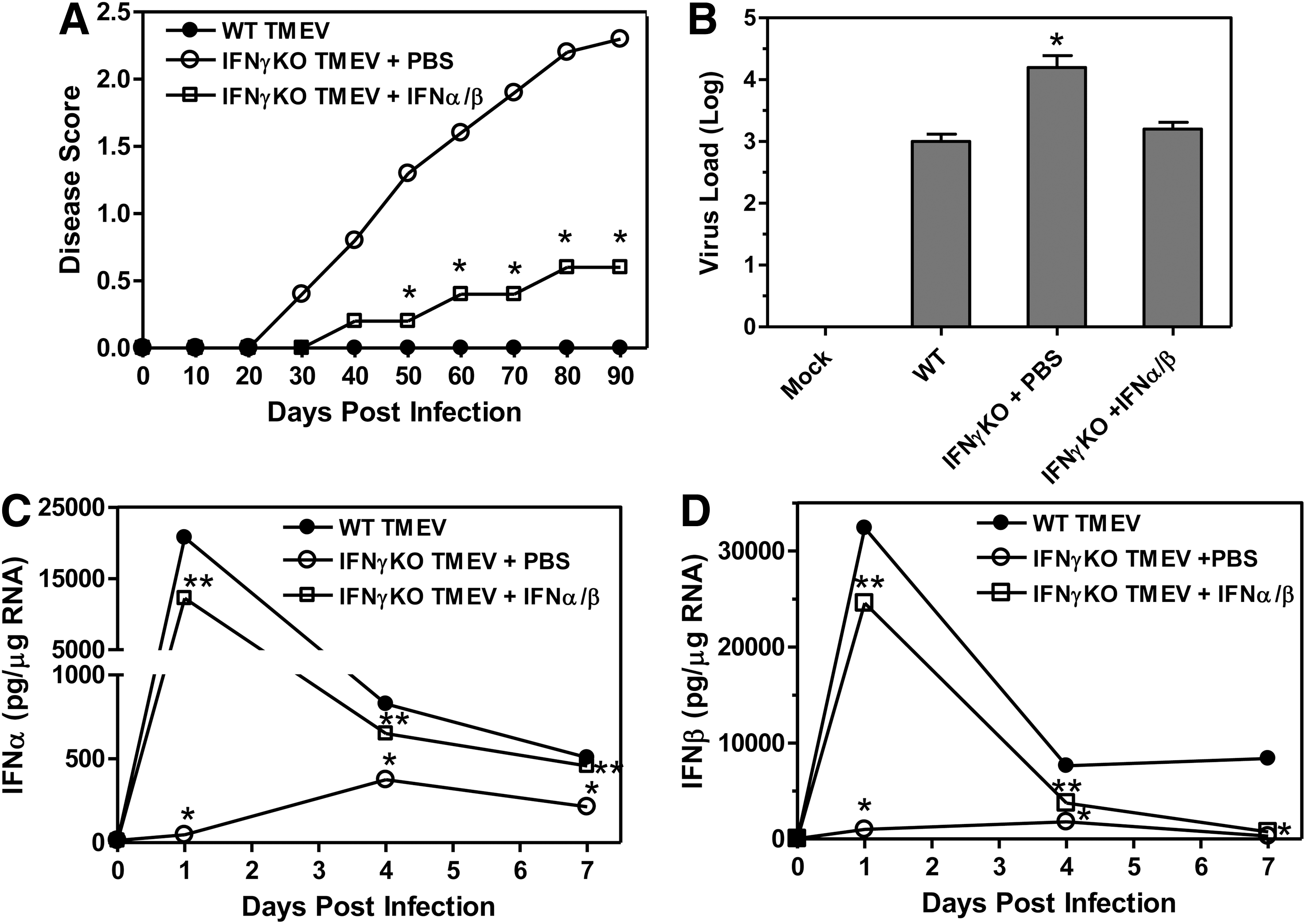
Administration of type I interferons to IFNγ-deficient mice during the innate immune response to TMEV reduced development of demyelinating disease. IFNγ-deficient mice were infected with TMEV and administered PBS or IFNα/ß by i.p. injection on the day of infection and day 4 post infection. The mice (10 mice per group) were monitored for clinical disease and scored based on a scale of 0–5
Next, we determined whether administration of type I interferons to IFNγ-deficient mice during the innate immune response to TMEV altered the expression of innate immune cytokines and chemokines. IFNγ-deficient mice were infected with TMEV and administered type I interferons or PBS (control) (3 mice per group), and the CNS was examined for expression of cytokines TNFα, IL-6, and IL-12 and chemokines, CXCL10 (IP-10) (Fig. 7). TMEV-infected IFNγ-deficient mice administered type I interferons had increased expression of TNFα, IL-6, and IL-12 compared to control treated mice. Likewise, TMEV-infected IFNγ-deficient mice administered type I interferons had increased expression of chemokines, CXCL10, compared to control treated mice. These results show that administration of type I interferons to IFNγ-deficient mice during TMEV infection increased the expression of innate immune cytokines and chemokines. Most interesting, the expression levels of TNFα, IL-6, IL-12, and CXCL10 in the IFNγ-deficient mice administered type I interferons during the innate immune response were similar to the levels observed in wild-type mice following TMEV infection (Fig. 7).
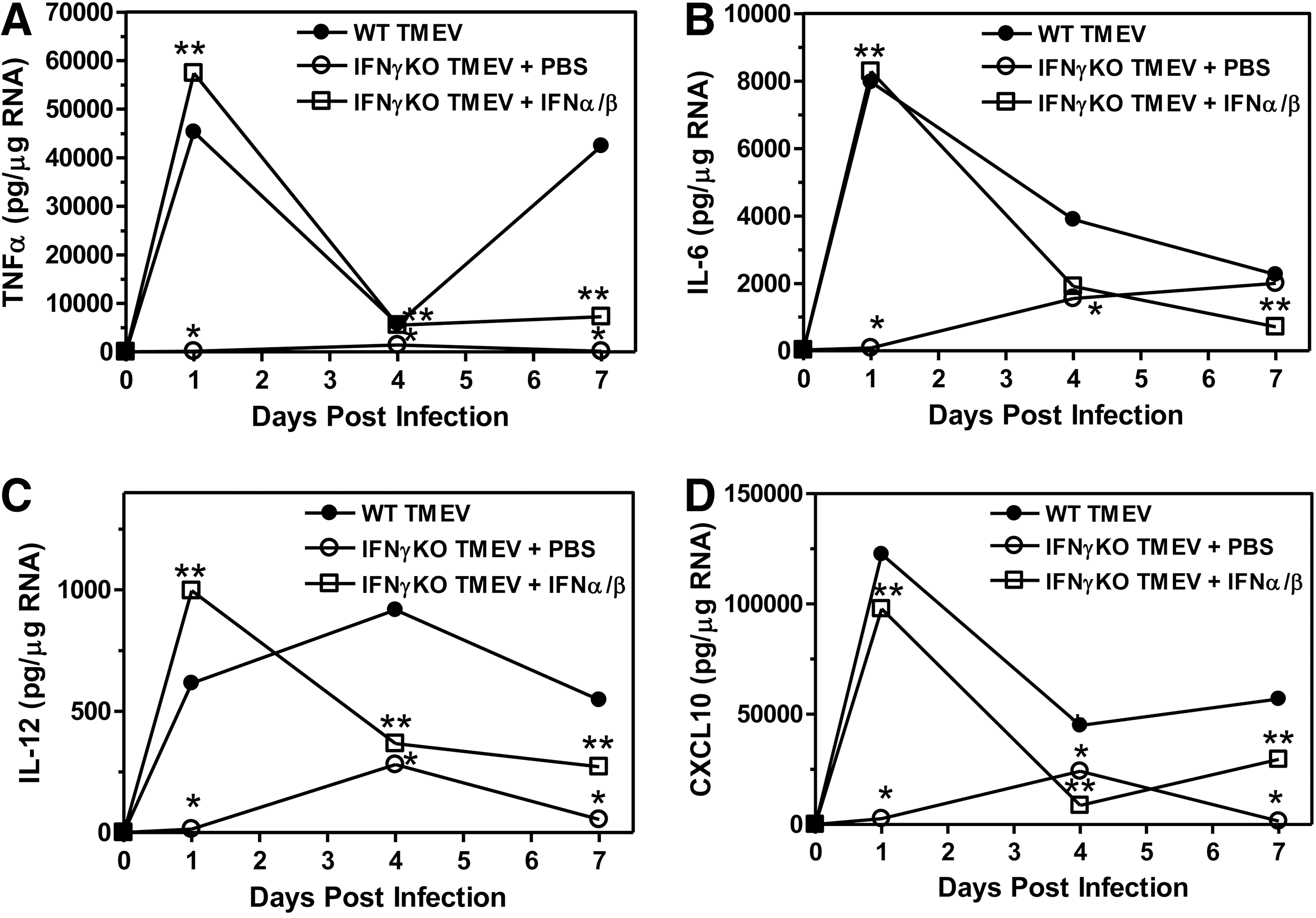
Administration of type I interferons to TMEV-infected IFNγ-deficient mice increased the expression of innate immune cytokines and chemokines in the CNS. Wild-type mice or IFNγ-deficient mice were infected with TMEV and administered PBS or IFNα/ß on the day of infection and day 4 post infection. The brain was removed from mice (3 mice per group) at 1, 4, and 7 days post infection. The RNA was isolated, DNAse treated, converted to cDNA, and real time PCR was conducted with primers for TNFα
As shown above, IFNγ-deficient mice had reduced infiltration of innate immune cells into the CNS following TMEV infection compared to wild-type mice. Further, administration of type I interferons to TMEV-infected IFNγ-deficient mice increased the expression of chemokines, such as CXCL10, during the innate immune response (shown above). Therefore, the immune cell infiltration into the CNS following administration of type I interfersons to TMEV-infected IFNγ-deficient mice (3 mice per group) was examined (Fig. 8). TMEV-infected IFNγ-deficient mice administered IFNα and IFNß had increased infiltration of macrophage compared to control treated mice (Fig. 8A). Similarly, TMEV-infected IFNγ-deficient mice administered type I interferons during the innate immune response had slightly increased infiltration of NK cells and increased infiltration of CD11b+Gr-1+ cells into the CNS compared to control treated mice (Fig. 8B). However, administration of type I interferons to TMEV-infected IFNγ-deficient mice did not significantly alter the infiltration of CD4+ and CD8+ T cells into the CNS (Fig. 8C). These results show that administration of type I interferons to IFNγ-deficient mice during the innate immune response to TMEV increased the infiltration of immune cells into the CNS to similar levels as wild-type mice (Fig. 8). Overall, these results show that IFNγ-deficient mice administered type I interferons following TMEV infection have an increased innate immune response that leads to reduced development and progression of demyelinating disease.
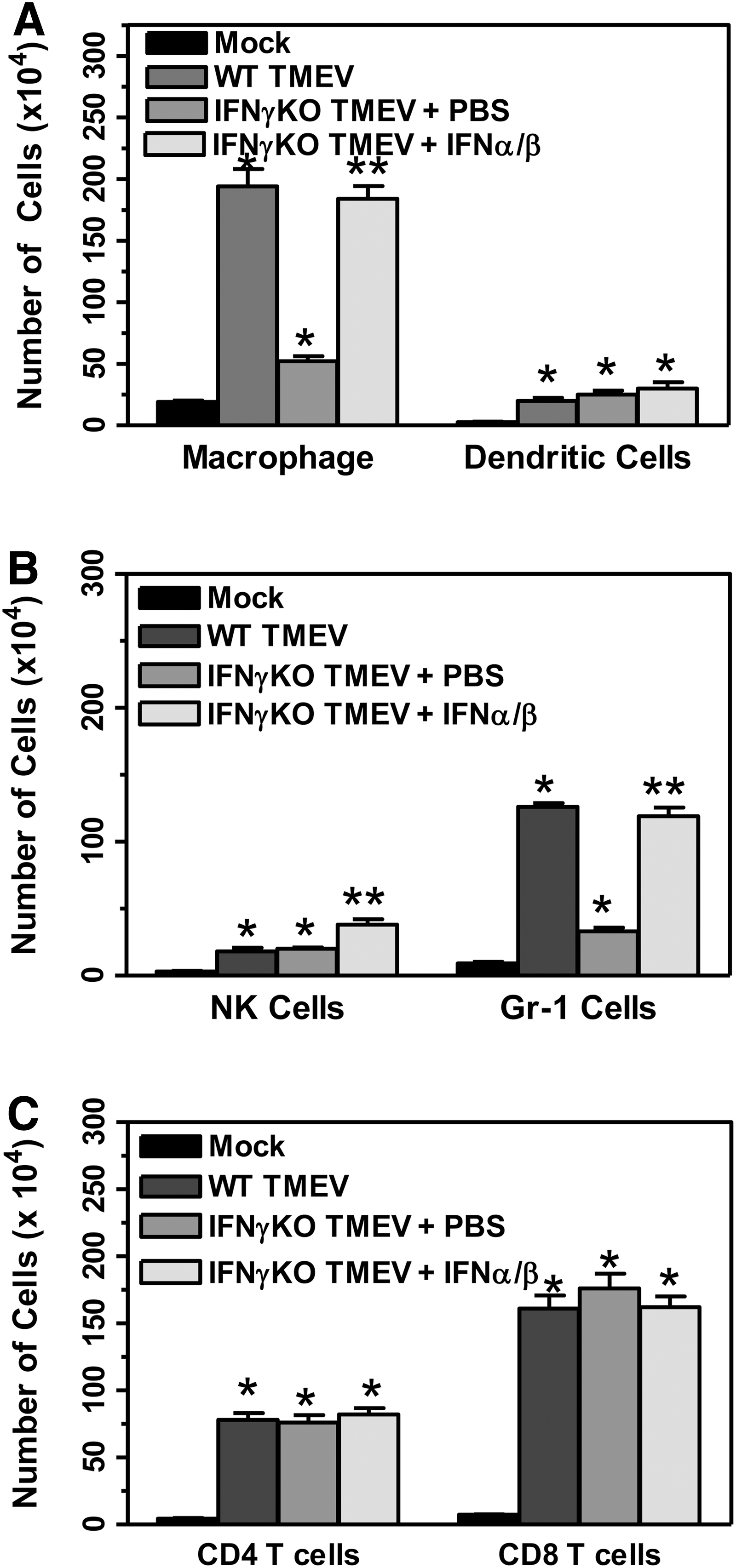
Administration of type I interferons to TMEV-infected IFNγ-deficient mice increased early immune cell infiltration into the CNS. IFNγ-deficient mice were infected with TMEV and administered PBS or IFNα/ß on days 0 and 4 post infection or were mock-infected. The brain and spinal cord were removed from mice (3 mice per group) at day 7 post infection, and the mononuclear cells were isolated. The cells were stained with fluorescently labeled antibodies for CD45, CD11b, CD11c, NK1.1, Gr-1, CD4, and CD8. The cells were analyzed by flow cytometry gated on CD45 high expressing cells. The number of macrophage (CD11b+CD11c- cells)
Microglia have a reduced innate immune response in IFNγ-deficient mice that can be increased by the administration of type I interferons after TMEV infection
Previous studies have suggested that type I interferons and type II interferon do not have redundant roles in the immune response against virus infection in the CNS (12). However, our results show that the lack of IFNγ has an effect on the innate immune response to TMEV, including reduced expression of type I interferons. Recent studies have shown that IFNγ has antiviral properties and has an effect on innate immune signaling (23,31). As shown above, IFNγ is expressed in the brain during the innate immune response to TMEV in wild-type C57BL6 mice (Fig. 2). Further, we have shown that IFNγ can activate microglia, the CNS resident macrophage population, to express pro-inflammatory cytokines and chemokines (32). We have also shown that microglia infected with TMEV express innate immune cytokines and chemokines. Therefore, we wanted to determine whether the effect of IFNγ on the innate immune response may be mediated through the activation of innate immune cells in the CNS, which are the microglia. IFNγ-deficient mice were infected with TMEV and administered IFNα/IFNß, IFNγ, or PBS (control) on the day of infection (8 mice per group). Microglia were isolated from the CNS of mice at day 2 post infection and analyzed for expression of innate immune cytokines, IFNα, IFNß, IL-6, IL-12, TNFα, and chemokine, CXCL10 (Fig. 9). Microglia isolated from IFNγ-deficient mice infected with TMEV had reduced expression of innate immune cytokines and chemokines compared to wild-type mice, which is similar to the results obtained from whole brain (Fig. 2). Administration of IFNα and IFNß to IFNγ-deficient mice infected with TMEV increased the expression of innate immune cytokines, including type I interferons, by microglia. Interestingly, administration of IFNγ to IFNγ-deficient mice infected with TMEV also increased the expression of innate immune cytokines by microglia returning the levels to near wild-type levels. Most interesting, administration of either IFNγ or IFNα and IFNß had a similar effect on microglia expression of innate immune cytokines and chemokines.

Administration of type I interferons or IFNγ to TMEV-infected IFNγ-deficient mice increased innate immune activation of microglia. Wild-type or IFNγ-deficient mice were infected with TMEV, and IFNγ-deficient mice were administered PBS, IFNα/ß or IFNγ on the day of infection. IFNγ-deficient mice were mock infected. The brain and spinal cord were removed from mice (8 mice per group) at day 2 post infection, and the mononuclear cells were isolated. The cells were stained with fluorescently labeled antibodies for CD45 and CD11b. The microglia were sorted based on CD45 intermediate CD11b+ cells. The cells were lysed and RNA was isolated and converted to cDNA for real time PCR with primers for IFNα
Innate immune cytokine expression is induced after infection through recognition of molecular patterns by innate immune receptors such as TLRs. We have previously shown that microglia express several TLRs and that expression of TLRs can be increased following IFNγ stimulation or TMEV infection of microglia (33). Thus, we wanted to determine whether IFNγ altered the expression of TLRs on microglia during TMEV infection (Fig. 10). Interestingly, microglia from TMEV-infected IFNγ-deficient mice had slightly reduced expression of TLR4 and TLR7 but not TLR3 compared to microglia from wild-type mice. TLR expression in the microglia could be returned to wild-type levels by the administration of either IFNγ or IFNα and IFNß. Furthermore, IFNγ has been shown to induce the expression of signaling molecule, IRF1, which can promote the transcription of type I interferons and innate immune cytokines (23). Interestingly, microglia from TMEV-infected IFNγ-deficient mice had reduced expression of IRF1 compared to wild-type mice and expression could be restored by the administration of IFNγ, but not by the administration of type I interferons. These results suggest that IFNγ plays an important role in enhancing activation of innate immune cells, such as microglia, through innate immune receptors and signaling molecules during TMEV infection. This innate immune activation is important for protecting the mice from establishing a persistent virus infection associated with development of demyelinating disease.
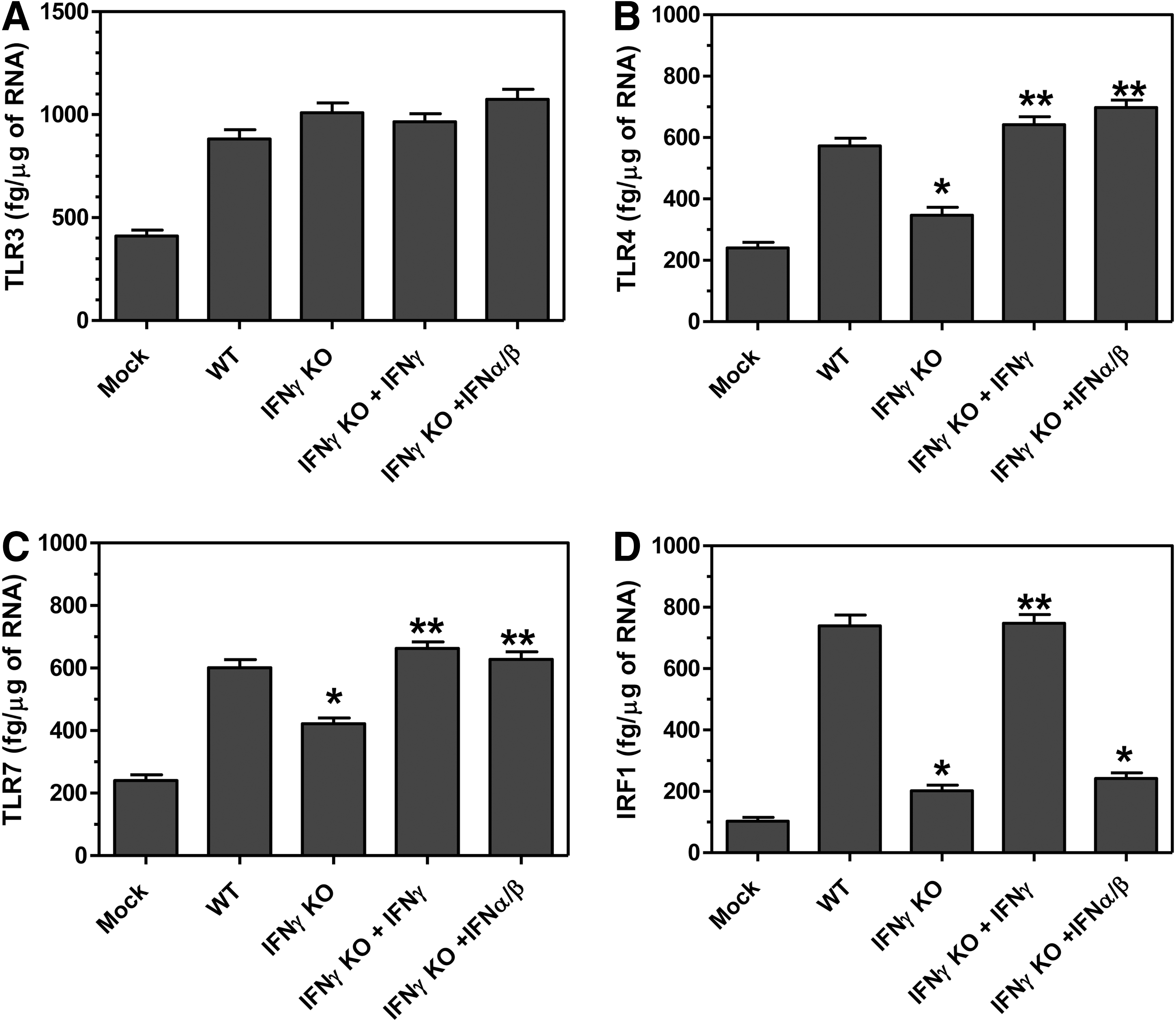
IFNγ alters innate immune receptor and signaling molecule expression in microglia during TMEV infection. Wild-type or IFNγ-deficient mice were infected with TMEV, and IFNγ-deficient mice were administered PBS, IFNα/ß or IFNγ on the day of infection. IFNγ-deficient mice were mock infected. The brain and spinal cord were removed from mice (8 mice per group) at day 2 post infection, and the mononuclear cells were isolated. The cells were stained with fluorescently labeled antibodies for CD45 and CD11b. The microglia were sorted based on CD45 intermediate CD11b+ cells. The cells were lysed and RNA was isolated and converted to cDNA for real time PCR with primers for TLR3
Discussion
We have shown that IFNγ-deficient C57BL6 mice infected with TMEV developed a demyelinating disease associated with an inflammatory immune response in the CNS similar to susceptible SJL mice. Most interesting, the TMEV-infected IFNγ-deficient mice had reduced expression of innate immune cytokines, especially type I interferons, and reduced infiltration of innate immune cells into the CNS, compared to wild-type mice. However, IFNγ-deficient mice developed a CD4+ and CD8+ T cell response against TMEV similar to wild-type mice. These results suggest that IFNγ-deficient mice have a reduced innate immune response to TMEV, which may contribute to the establishment of a persistent virus infection in the CNS and development of demyelinating disease. Therefore, we administered type I interferons to IFNγ-deficient mice during the innate immune response to TMEV to compensate for the deficit in the innate immune response. IFNγ-deficient mice administered type I interferons during the innate immune response to TMEV had an increased innate immune response, including increased expression of innate immune cytokines and increased infiltration of innate immune cells into the CNS. Most importantly, IFNγ-deficient mice administered type I interferons during the innate immune response to TMEV had reduced development and progression of demyelinating disease. These results suggest that IFNγ plays an important role in promoting the innate immune response that protects mice from development of TMEV-induced demyelinating disease.
IFNγ can be secreted from a variety of different cells and has several immunomodulatory functions. IFNγ is secreted by Th1 type CD4+ T cells and promotes Th1 type CD4+ T cells while suppressing Th2 type CD4+ T cells (13,38). In our studies, IFNγ-deficient mice infected with TMEV developed virus-specific CD4+ T cell response similar to wild-type mice. IFNγ can also be secreted by CD8+ T cells and has been suggested to mediate noncytolytic functions involved in clearance of virus infections from the CNS (24,36). In our studies, IFNγ-deficient mice infected with TMEV developed virus-specific CD8+ T cells with similar lytic functions as wild-type mice, however we did not determine whether nonlytic functions of CD8+ T cells were affected in the IFNγ-deficient mice. IFNγ secreted by T cells, especially CD4+ T cells, activates macrophage to express cytokines and chemokines, and activates macrophage to upregulate MHC class II and co-stimulatory molecules for antigen presentation. IFNγ has also been shown to enhance dendritic cell functions by upregulating expression of MHC class II and by increasing expression of IL-12 which promotes the Th1 type CD4+ T cell response (1,27). Furthermore, IFNγ has been shown to activate microglia to increase expression of proinflammatory cytokines and upregulate expression of MHC class II and co-stimulatory molecules for antigen presentation (32). The results from the current studies showed that IFNγ-deficient mice had reduced macrophage infiltration into the CNS during the innate immune response, however macrophage and microglia isolated from the CNS of TMEV-infected IFNγ-deficient mice expressed similar levels of MHC class II and co-stimulatory molecules as cells from wild-type mice (data not shown). Therefore, these results suggest that T cells and antigen presenting cells were able to be activated in the absence of IFNγ during TMEV infection.
IFNγ is secreted by Th1 type CD4+ T cells associated with autoimmune demyelinating disease, and secretion of IFNγ has been correlated with disease severity (17). However, studies in the EAE model of autoimmune demyelinating disease have shown that IFNγ may be playing a protective role during disease. Mice that are normally resistant to EAE induction become susceptible to demyelinating disease in the absence of IFNγ, and susceptible mice develop a more severe demyelinating disease in the absence of IFNγ (11,16). Additional studies have demonstrated that expression of IFNγ may determine the localization of lesions during EAE. In the absence of IFNγ, mice develop lesions in the brain stem and cerebellum compared to wild-type mice which develop lesions in the spinal cord (20). IFNγ has also been shown to be protective following TMEV infection of the CNS by preventing lethal infection of anterior horn neurons and primary spinal motor neurons (37). In the current studies, we showed that resistant mice that lack IFNγ develop demyelinating disease similar to susceptible mice with similar clinical symptoms and inflammatory response as susceptible mice. We have further shown that the T cell response to the virus continues throughout the persistent virus infection in TMEV-infected IFNγ-deficient mice and that T cells may be involved in the inflammatory response during demyelinating disease, which suggests the lack of IFNγ may not affect activation of pathogenic T cells. These results suggest that IFNγ may be mediating protection through other immune cells or through CNS resident cells.
Recent studies have suggested a role for IFNγ in the innate immune response. Innate immune receptors, such as TLR3, TLR7, and MDA5, recognize the pathogen associated molecular pattern of double-stranded RNA during virus infections, such as TMEV. A recent study examining the innate immune response to coxsackievirus B3 determined that recognition of dsRNA by TLR3 leads to the expression of IFNγ and recognition of dsRNA by MDA5 receptor leads to expression of type I interferons (31). The study further demonstrated that the TLR3–IFNγ pathway was critically important in the innate immune response, and that together with MDA5- type I interferon pathway was required to mount a protective antiviral response. Thus, type I and type II interferons (IFNγ) work in parallel to successfully mount an innate immune response to a virus infection. In the current studies, mice deficient in IFNγ had reduced innate immune response following TMEV infection. Microglia from IFNγ-deficient mice had similar TLR3 expression as wild-type mice but reduced expression of TLR4 and TLR7.
Type I interferons, IFNα and IFNß, bind to a common receptor, IFNα/ß receptor, and type II interferon, IFNγ, binds to IFNγ receptor. Interestingly, both receptors lead to phosphorylation and activation of signaling molecules that then translocate to the nucleus to activate transcription of genes involved in host defense. Studies have demonstrated that IFNγ strongly augments type I interferon response by increasing that activation of signaling molecules (8). Likewise, type I interferon can increase the amount of signaling molecule, STAT1, present in cells which boosts the signaling response upon IFNγ receptor binding (43). Both type I and type II interferons induce the expression of IFN-stimulated genes (ISGs), which have a variety of functions from direct antiviral activity to activation of other signaling molecules (9). IRF1 is an ISG that promotes the expression of type I interferons (23,30). IFNγ has been shown to induce high levels of IRF1 which promotes the expression of type I interferons and innate immune cytokines, thus an important antiviral mechanism of IFNγ may be to induce IRF1 creating a positive feedback loop for the expression of type I interferons (23). IRF1 has also been shown to associate with TLR signaling molecule MyD88 (30). The IRF1 associated with MyD88 was more efficient at migrating to the nucleus and inducing gene expression, which suggests that IFNγ enhances TLR signaling and expression of innate immune cytokines (30). In addition, the protective innate immune response to encephalomyocarditis virus (EMCV) has been shown to be dependent on IFNγ-mediated IRF1 induction (15). Our studies show that microglia from IFNγ-deficient mice had reduced expression of innate immune cytokines and that microglia from IFNγ-deficient had reduced IRF1 expression, thus, IFNγ may be activating IRF1 following TMEV infection, which enhances the expression of type I interferons and innate immune cytokines. The addition of IFNα and IFNß did not significantly affect IRF1 expression but was able to restore the innate immune response in microglia from IFNγ-deficient mice, which suggests that type I interferons increase the expression of innate immune cytokines using other signaling molecules.
We have previously shown that IFNγ can stimulate microglia to express low levels of IFNß and that TMEV infection can stimulate microglia to express large amounts of IFNα and IFNß, as well as other innate immune cytokines (32). We have also shown that microglia express all the TLRs and upregulate TLR expression upon TMEV infection or IFNγ stimulation. In the current studies, we examined microglia isolated directly from IFNγ-deficient mice infected with TMEV. Microglia from TMEV-infected IFNγ-deficient mice had reduced expression of type I interferons, IFNα and IFNß, and other innate immune cytokines compared to microglia from wild-type mice. Administration of type I interferons or IFNγ to IFNγ-deficient mice increased expression of type I interferons by microglia, which suggests that IFNγ may play an important role in enhancing the expression of type I interferons by microglia in the CNS during the innate immune response to TMEV. Next, we wanted to determine whether IFNγ was altering the receptors and signaling molecules involved in the innate immune response. Interestingly, microglia from TMEV-infected IFNγ-deficient mice had reduced expression of TLR4 and TLR7 but not TLR3, suggesting that IFNγ may be enhancing TLR expression during virus infection through MyD88 dependent pathway. Furthermore, microglia from TMEV-infected IFNγ-deficient mice had reduced expression of IRF1 which is induced by IFNγ and promotes the expression of innate immune cytokines. Previous studies have suggested that IRF1 associates with MyD88 to enhance signaling during the innate immune response (30). Therefore, IFNγ influences the expression of innate immune cytokines through increasing expression of innate immune receptors and signaling molecules.
Immediately after infection, type I interferons are expressed through signaling from the innate immune receptors. The type I interferons are secreted from the cells and bind to the IFNα/ß receptor, which creates a feed-back loop that drives the expression of type I interferons during the innate immune response. In the absence of IFNγ, signaling through the IFNγ receptor would be eliminated reducing the amount of transcription factors, such as IRF1, which promotes the expression of IFNα and IFNß. Therefore, IFNγ may be cooperating with type I interferons through activating signaling molecules to induce the expression of cytokines during the innate immune response. In the current studies, the innate immune response could be restored in IFNγ-deficient mice by the administration of type I interferons, which suggests that increasing the amount of type I interferon present during the innate immune response could overcome the deficit produced by the lack of IFNγ. Type I interferons play a very important role in the innate immune response to TMEV. Mice deficient in IFNα/ß receptor are very susceptible to TMEV infection and die very early following infection (data not shown) (12). We have recently shown in susceptible mice that reducing the amount of IFNß present during the innate immune response to TMEV leads to development of a more severe demyelinating disease while increasing the amount of IFNß present during the innate immune response reduces the development of demyelinating disease (34). Therefore, the amount of IFNß expressed during the innate immune response to TMEV affects the development and progression of demyelinating disease.
The current studies show that TMEV infection of resistant mice deficient in IFNγ leads to a reduced innate immune response and development of demyelinating disease. Administration of type I interferons during the innate immune response restores the innate immune response and reduces the development of demyelinating disease in IFNγ deficient mice. These results suggest that IFNγ may be boosting the innate immune response by enhancing the activation of signaling molecules in innate immune cells, such as microglia, which secrete type I interferons and innate immune cytokines. The effective innate immune response enables control of virus replication and promotes an adaptive immune response that clears the virus from the CNS and protects C57LB6 mice from developing demyelinating disease.
Footnotes
Acknowledgments
This work was supported by a grant from the National Multiple Sclerosis Society, RG3625. We thank Emily Thompson, Erin Johnson, and Samuel Hall at the University of Wisconsin for technical assistance.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.