
Abstract
Infection with influenza A virus (IAV) leads to acute lung injury and possibly fatal complications, especially in immunocompromised, elderly, or chronically infected individuals. Therefore, it is important to study the factors that lead to pathology and mortality in infected hosts. In this report, we analyze immune responses to infection at a sublethal (0.1 LD50) and lethal (1 LD50) dose of the highly pathogenic IAV A/Puerto Rico/8/34 (PR8). Our experiments revealed that infection with a 1 LD50 dose induced peak viral titers at day 2 compared to day 4 in the 0.1 LD50 dose. Moreover, early cytokine dysregulation was observed in the lethal dose with significantly elevated levels of IFN-α, TNF-α, CXCL9, IL-6, and MCP-1 produced at day 2. Early inflammatory responses following infection with 1 LD50 correlated with a greater influx of neutrophils into the lung. However, depletion of neutrophils enhanced morbidity following IAV infection. Though no differences in CD8+ cell function were observed, CD4+ effector responses were impaired in the lungs 8 days after infection with 1 LD50. Histological analysis revealed significant pathology in lethally infected mice at day 2 and day 6 postinfection, when viral titers remained high. Treating lethally infected mice with oseltamivir inhibited viral titers to sublethal levels, and abrogated the pathology associated with the lethal dose. Together, these results suggest that early cytokine dysregulation and viral replication play a role in pulmonary damage and high mortality in lethally infected mice.
Introduction
E
IAV is a negative stranded enveloped RNA virus that productively infects and replicates within airway epithelial cells in the lung respiratory tract. Following infection, viral nucleic acids bind to pattern recognition receptors (PRR) within epithelial and immune cells (32) causing high levels of inflammatory cytokines, including TNF-α, IL-6, IL-1α/β, IFN α/β, CXCL9/10, MIP-1α/β, and MCP-1 (27), to be secreted in the lung. The presence of these cytokines, and others, alters the lung microenvironment and initiates the trafficking of immune cells such as macrophages, dendritic cells, and neutrophils to the site of infection. Subsequently, antigen presenting cells (APC) migrate back to the draining lymph nodes to activate adaptive immune cells that are required for clearing IAV infection. The generation of antigen specific CD4+ and CD8+ cells are not only directly responsible for viral clearance, but are correlated with enhanced protection from IAV (2,10,14,20,21,40).
While robust immune responses are critical for IAV clearance, unintended immunopathology has been attributed to destruction of tissues within the lung and increased morbidity (5,23). Due to the pathogenic nature of pandemic IAV infections, a number of reports have associated the “cytokine storm” as the primary cause of mortality in infected hosts (3,9,15,38). This paradigm states that highly virulent IAV strains induce elevated cytokine levels that synergistically promote a pathological inflammatory environment responsible for severe disease in infected hosts. It is not clear whether abrogating the cytokine storm in IAV infections can prevent host mortality. However, several studies have examined the connection between cytokine levels and immunopathology following IAV infection. Experiments with knockout mice have shown that eliminating specific cytokines from the immune response such as IL-1β (39), TNF-α (28), MCP-1 (28), IL-6 (28,34), and MIP-1α (34) does not rescue the lethal phenotype. Additional reports have used IAV strains with varying degrees of virulence to assess immune responses and have found that more virulent viruses induce higher levels of cytokines and decreased survival (18). Despite recent studies addressing the cytokine storm with virulent IAV strains or knockout mice, little has been done to examine this phenomenon using the same virus isolate, an approach where the only difference between experimental groups is the viral inoculum. Here, we sought to identify differential cytokine profiles and therapeutic depletion strategies to enhance survival in lethally infected mice.
In this study, we use a model of acute IAV infection to outline and compare various immune parameters following infection with either a sublethal or a lethal dose of PR8 virus. We found that administration of a lethal dose of IAV increased levels of PRRs and cytokines at early time points. Additionally, infection with a lethal dose led to robust influx of neutrophils on day 2, but depleting them from the response led to enhanced morbidity. Paradoxically, the increased levels of inflammatory mediators early in the response had no effect on the number of T-cells entering the lung. However, CD4+ Granzyme B (GrB) mean fluorescence intensity (MFI) and IFN-γ secretion were diminished in mice receiving the lethal dose, but CD8+ T-cell responses were unchanged. Furthermore, the pathology associated with a high infectious dose was significantly higher at days 2 and 6 postinfection compared to sublethally infected mice. To rescue the lethal phenotype, mice were treated with oseltamivir, and reduced levels of immunopathology and viral load were observed. These findings suggest that the pathology and mortality associated with IAV infection depends on the initial viral inoculum and early viral replication, but not necessarily on high cytokine expression early after infection.
Materials and Methods
Mice
Male BALB/c mice were purchased from Jackson Laboratories (Bar Harbor, ME). Mice 6–8 weeks old were used in all experiments. Experimental animal procedures using mice were approved by and conducted in accordance with the Institutional Animal Care and Use Committee (IACUC) at the University of Nebraska–Lincoln.
Influenza virus infection
Mice were anesthetized with isoflurane and 30 μL PR8 influenza virus diluted in phosphate-buffered saline (PBS) was administered intranasally at a sublethal dose of 0.1 LD50 or at a lethal dose of 1, 1.5, or 2 LD50. Weight loss was monitored, and survival curves were generated.
Extraction of RNA and real-time quantitative reverse transcription polymerase chain reaction
Mice were sacrificed every 2 days following intranasal infection, and the lungs were placed immediately in RNAlater (Ambion, Austin, TX) and then frozen at −20°C. The samples were weighed and homogenized in TRIzol (Ambion) at 1 mL/100 mg of lung tissue using a Tissue Tearor homogenizer (Biospec Products, Inc., Bartlesville, OK). RNA was isolated from lung homogenates using RiboPure RNA kit (Ambion): 2 μg of RNA was reverse transcribed into cDNA using a high capacity cDNA Reverse Transcription kit as per the manufacturer's protocol (Applied Biosystems, Carlsbad, CA). Then 50 ng of cDNA was used for amplification by quantitative real-time PCR (Step One Plus; Applied Biosystems). Specific primers for murine TLR3 (Mm01207403_m1), TLR7 (Mm00446590_m1), TLR9 (Mm00446193_m1), Ddx58 (RIG-I) (Mm00554529_m1), TNF-α (Mm00443258_m1), MIP-1β (Mm00443111_m1), and IL-6 (Mm0044619_m1) were purchased from Applied Biosystems. The following murine primers were purchased from Integrated DNA Technologies (Coralville, IA). IFN-β1: 5′-/56-FAM/TCC ACT CTG/ ZEN/ ACT ATG GTC CAG GCA/ 3IA BkF Q/-3 (probe); 5′-GGC TAG GAG ATC TTC AGT TTC G-3′ (forward); 5′-AGG ATT CTG CAT TAC CTG AAG G-3′ (reverse). IFN-α4: 5′-/56-FAM/TTT GGA TTC/ ZEN/ CCC TTG GAG AAG GTG G/3I ABK FQ/-3′ (probe); 5′-GCC TTC TGG ATC TGT TGG TTA-3′ (forward); 5′-GCC TCA CAC TTA TAA CCT CGG-3′ (reverse). CXCL9: 5′-/56-FAM/ CTG GAG CAG/ ZEN/ TGT GGA GTT CGA GG/3 IAB kFQ/-3′ (probe); 5′-TGA GGT CTT TGA GGG ATT TGT AG-3′ (forward); 5′-AGT CCG CTG TTC TTT TCC TC-3′ (reverse).
To determine the viral titer, the following acid polymerase (PA) probe and primers were used: 5′-/56-FAM/CCA AGT CAT/ ZEN/ GAA GGA GAG GGA ATA CCG CT/3 IAB kFQ/-3′ (probe), 5′-CGG TCC AAA TTC CTG CTG AT-3′ (forward), 5′-CAT TGG GTT CCT TCC ATC CA-3′ (reverse). A known concentration of PA-containing plasmid was used to generate a standard curve in all reactions. PA copies per lung were then calculated based an initial concentration of 50 ng of cDNA as described (14).
Mouse multiplex assay
Mice were infected as described previously. At 2-day intervals following infection, mice were sacrificed, and bronchoalveolar lavage (BAL) fluid was collected. A BD cytometric bead array (CBA) was performed on the BAL samples according to the manufacturer's protocol and acquired using a BD FACSArray. Cytokines were calculated from a standard curve using FCAP Array Software (from BD Biosciences, San Jose, CA).
Isolation of lung and lymph node cells for flow cytometry
Mice were sacrificed at various times postinfection with a lethal dose of tribromoethanol (Avertin) intraperitoneally. Lungs and draining lymph node cells (a pool of mediastinal and cervical lymph nodes) were processed as described (2) for flow cytometry analysis. Briefly, lungs were perfused with PBS, treated with collagenase D, and filtered through a 70 μm filter. Additionally, draining lymph node cells were dissociated into single cell suspensions using 70 μm filters. Cell suspensions were stained with fluorochrome-conjugated antibodies to anti-CD4, CD8, CD43, pan NK1.1 (CD49b), Gr-1, CD11c, and CD11b (eBioscience, Inc., San Diego, CA) for 30 min at 4°C. After washes, cells were resuspended in 500 μL of PBS containing 1% BSA and 0.1% NaN3. In some experiments, T-cells were surface stained, fixed in 1% paraformaldehyde, and stained with anti-GrB (Invitrogen, Carlsbad, CA) antibody to measure intracellular levels of GrB protein in effector T-cells. Cells were acquired using a FACS Calibur flow cytometer (BD Biosciences) and analyzed using FlowJo software (Tree Star, Inc., Ashland, OR).
Restimulation with peptides for cytokine analysis
For intracellular cytokine assays, cells were isolated from the lungs as described above, and restimulated with influenza peptide pulsed A20s as APCs in RPMI 1640 containing 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mM
Antibody-mediated neutrophil depletion
Anti-Ly6G (RB6-8C5) and IgG2b (LTF-2) antibodies were purchased from BioXcell (West Lebanon, NH). For neutrophil depletion, mice were treated on day −1 or day 1 postinfection with 1 mg/mouse of RB6-8C5 or IgG2b isotype control antibody by intraperitoneal injection. Mice were infected with 1 LD50 PR8, and survival and weight loss were monitored.
In vivo administration of oseltamivir
Mice were anesthetized with isoflurane and infected intranasally with either 0.1 LD50, 1.5 LD50 of PR8, or mock infected with PBS. Oseltamivir was administered by oral gavage twice daily for 5 days at 10 mg/kg/day (41). Mice that were mock infected with PBS were given oseltamivir on the same schedule. Four hours after the first dose of oseltamivir, mice were infected with IAV, and weight loss was monitored for 8 days. After 8 days, mice were sacrificed and lungs were processed for histological evaluation.
Histopathology
Mice were sacrificed using Avertin as described, and lungs were insufflated with 10% neutral buffered formalin and immersed in the same solution for fixation. Following fixation, a cross-section of the right superior, right middle, right inferior, post-caval, and cross-sections from the cranial, middle, and caudal sections of the left lung were collected and routinely processed (Tissue-Tek VIP5 processor) for histological evaluation. Paraffinized sections were cut 4 μm thick and were stained with hematoxylin and eosin (Symphony stainer H&E stainer, Ventana, Tucson, AZ) for histological evaluation. The samples were randomized and graded by a blinded veterinary pathologist using a published grading scale (6). A grade of 0 was assigned to normal lungs; grade 1 connoted mild inflammation affecting <10% of the parenchyma; grade 2 connoted moderate lesions including inflammation of between 10% and 30% of the lung with moderate epithelial hyperplasia and necrosis; and grade 3 connoted severe lesions. In some experiments, the grading scale was expanded to account for subtle differences in the lung pathology. In addition to the whole numbers used by McAuley et al. (6), half values were added to the scale.
Statistics
Statistical significance between experimental groups was determined by two-tailed Student's t-test using Prism 4.0 (GraphPad Software). Survival curves were analyzed using the Kaplan–Meier method.
Results
0.1 LD50 and 1 LD50 infections differ in pathogenicity in BALB/c mice
Many studies have examined the pathogenicity and virulence factors implicated in morbidity induced by different strains of IAV viruses, yet little has been done to determine the immunological parameters that lead to mortality in mice using the same viral strain. To address this issue, we employed a model system that examines the immune response following infection with PR8 at a low (0.1 LD50) or a high (1 LD50) infectious dose. Mice infected intranasally with a 0.1 LD50 dose of IAV PR8 showed slight increases in weight 2 days postinfection, followed by steady weight loss until day 10 (Fig. 1A). Likewise, mice infected with 1 LD50 followed similar weight-loss kinetics, except to a more severe degree, losing on average 30% of their initial body weight. To confirm the lethality of the infectious doses used in this study, mice were infected and monitored for survival. All of the mice given a 0.1 LD50 survived, while half of the mice given a 1 LD50 succumbed to infection (Fig. 1B). To assess viral load in infected hosts, lung samples were tested for the presence of the IAV PA gene by quantitative reverse transcription polymerase chain reaction (qRT-PCR) (Fig. 1C). Throughout the time course, the 1 LD50 infection resulted in higher viral titers than infection with the sublethal dose. Furthermore, peak viral load was different with titers peaking at day 2 in the 1 LD50 infection and day 4 for the 0.1 LD50. By day 10, virus was still present in the 1 LD50 group, whereas there was no detectable level of virus in the lung in the 0.1 LD50 group. These data suggest that underlying immune factors could be responsible for differential viral clearance rather than viral virulence factors.
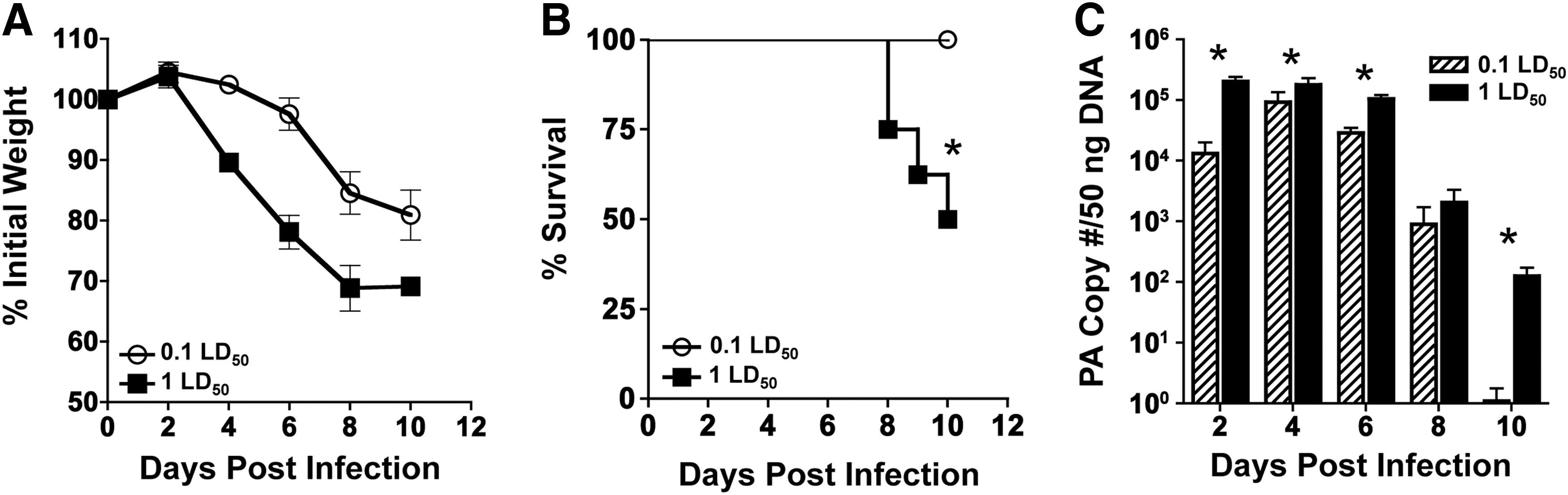
Sublethal (0.1 LD50) and lethal (1 LD50) infections differ in pathogenicity in BALB/c mice.
PRR expression and antiviral cytokines diverge at early time points in infected mice
IAV infection has been shown to activate a number of PRRs within infected lungs. To examine the early recognition of virus in the lung, a panel of PRRs implicated in infection was assessed by qRT-PCR (Fig. 2A). In all PRRs assessed, the 1 LD50 infection resulted in increased PRR expression over the sublethal dose at day 2. By day 4, PRR expression in the 0.1 LD50 reached similar expression levels as the 1 LD50 dose and maintained the same or higher levels of mRNA transcripts for the duration of the infection. Surprisingly, TLR9 was the most highly upregulated of any PRR tested. As expected, expression of PRRs tapered off by day 8 as the immune response transitioned from the innate to the adaptive response.
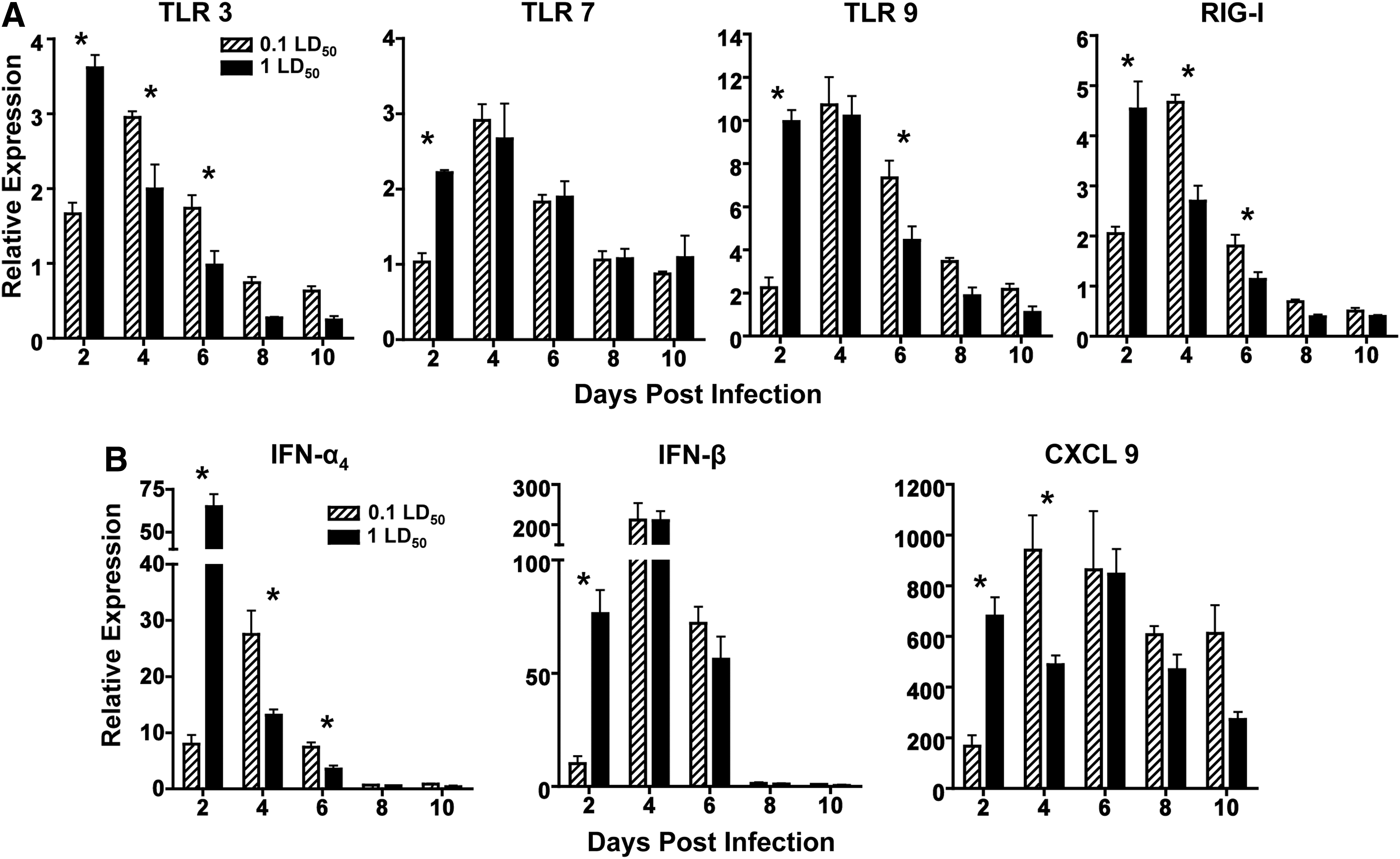
PRR expression and antiviral cytokines diverge at early time points in infected mice.
We observed similar trends in expression of other innate antiviral mediators as we observed in PRR expression. At day 2, infection with 1 LD50 resulted in higher levels of IFN-α4, IFN-β, and CXCL9 compared to 0.1 LD50 (Fig. 2B). By day 4, IFN-α4 and CXCL9 levels in mice infected with 0.1 LD50 surpassed that of the response during 1 LD50 infection. Additionally, the relative expression of IFN-β was much higher than IFN-α4 levels on days 4 and 6. At each time point, CXCL9 was upregulated, and its expression was sustained throughout the infection. These results suggest that the peak induction of the PRRs and innate cytokines correlate with peak of viral titers presumably leading to downstream effects including increased disease and mortality.
Inflammatory cytokines and chemokines are higher at day 2 in lethally infected mice
To assess the proteins generated in the inflammatory response further, we examined a number of critical antiviral and immunostimulatory cytokines present in the airways of infected mice. Cytokines involved in chemotaxis, shaping the adaptive response, and inflammation were examined. Mice were sacrificed at the indicated times post infection, and BAL fluid was collected for multiplex analysis. At day 2 postinfection, the 1 LD50 dose elicited higher concentrations of all cytokines tested compared to 0.1 LD50, with significant differences observed in IL-6, TNF-α, and MIP-1β (Fig. 3). In addition to the induction of cytokines at day 2, a second wave of inflammation was observed at day 6 after infection with 1 LD50, as demonstrated by increases in IL-6 and MCP-1 levels. In the cases of TNF-α, MIP-1α, and MIP-1β, the early differences in cytokine levels between the doses were resolved by day 4 (Fig. 3). These results further support the findings that infection with a higher dose of IAV leads to a higher inflammatory state in the lung at early time points.
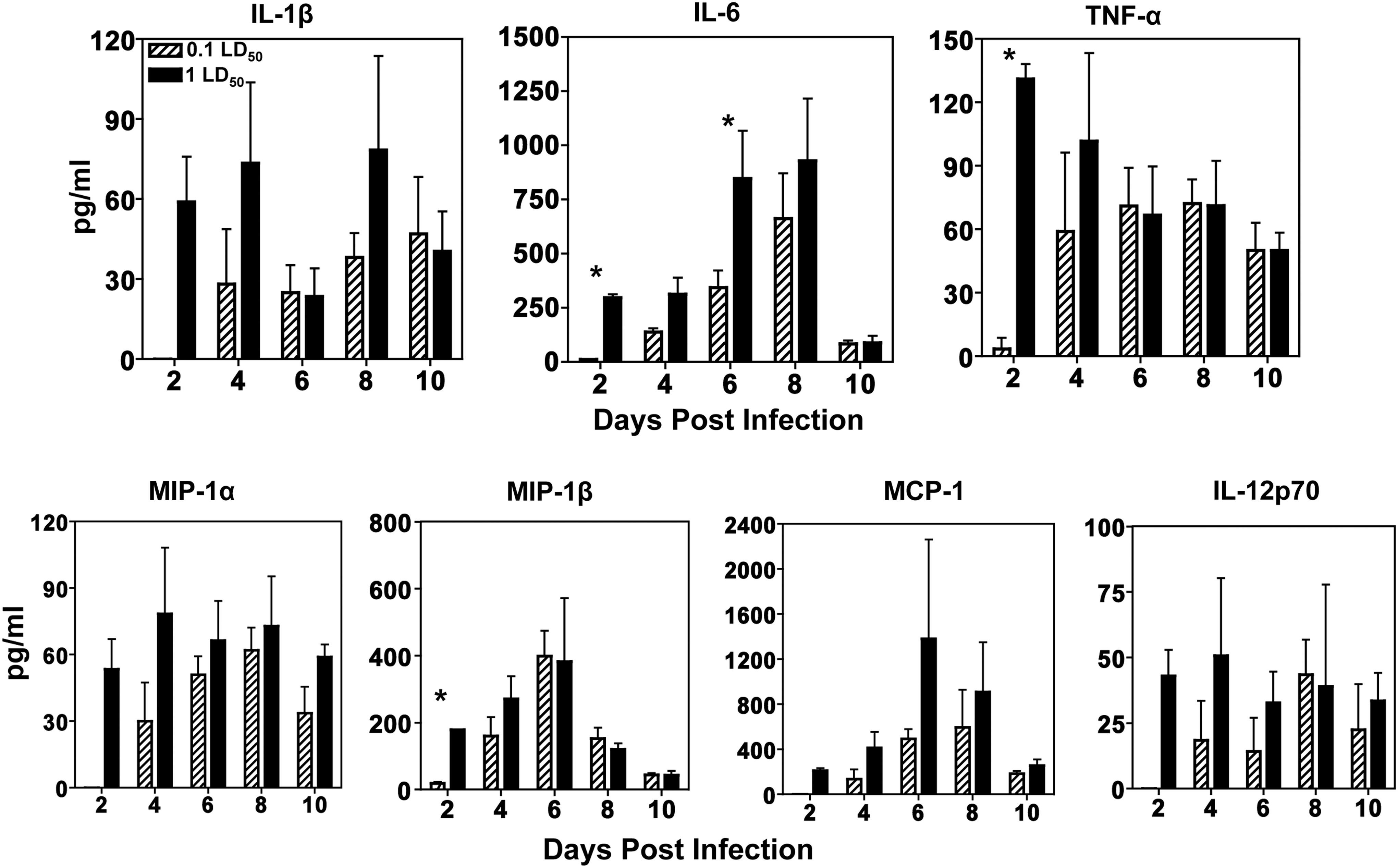
Inflammatory cytokines and chemokines are higher at day 2 in lethally infected mice. Groups of four to nine mice were infected as described previously. Bronchoalveolar lavage (BAL) fluid was collected from infected animals and subjected to a bead-based immunoassay as described in the Materials and Methods to measure various levels cytokines in the airways. Levels of all cytokines are shown as pg/mL of BAL fluid. A Student's t-test was used to calculate p-values (*p<0.05).
Blocking neutrophils enhances morbidity following influenza infection
Because the magnitude and phenotype of infiltrating leukocytes may be important indicators of mortality, cellular infiltrates from infected mice were measured. Since we observed an inflammatory state with increased cytokines in the airways after 1 LD50 infection, we expected corresponding differences in cellular influx in the airways. As seen in Figure 4A, approximately 40% of cellular infiltrates in the sublethal dose were CD11c+/CD11blo cells, while infection with a 1 LD50 induced the influx of more than 50% Gr-1hi/CD11b+ neutrophils. Following infection, mice given 1 LD50 had more than twice the number of Gr-1+/CD11b+ cells in the airways at day 2, yet no significant difference in CD11c+ cells were observed in the airways (Fig. 4B). Continuing from day 4 on, the total number of CD11b+/Gr-1+ cells in the mice infected with a sublethal dose reached similar levels as seen during infection with a lethal dose, and remained equivalent throughout the duration of infection. Thus, infection with a 1 LD50 generates more neutrophils and fewer CD11c+ cells overall at early time points compared to 0.1 LD50. Because of these early differences in infiltrating Gr-1+/CD11b+ cells, we hypothesized that depleting neutrophils would abrogate the lethal phenotype. Using a depleting antibody RB6-8C5, neutrophils were depleted for up to 4 days postinfection (data not shown). This strategy was employed to reduce the number of neutrophils in the lung early after infection as a potential therapy, with the expectation that neutrophils would return later in the response. Mice infected with IAV following neutrophil depletion showed enhanced morbidity and lost nearly 30% of their initial body weight compared to the 25% weight loss in mice receiving the isotype control antibody (Fig. 4C). This finding was in agreement with recent studies showing that neutrophils were necessary for survival from IAV infection (35).
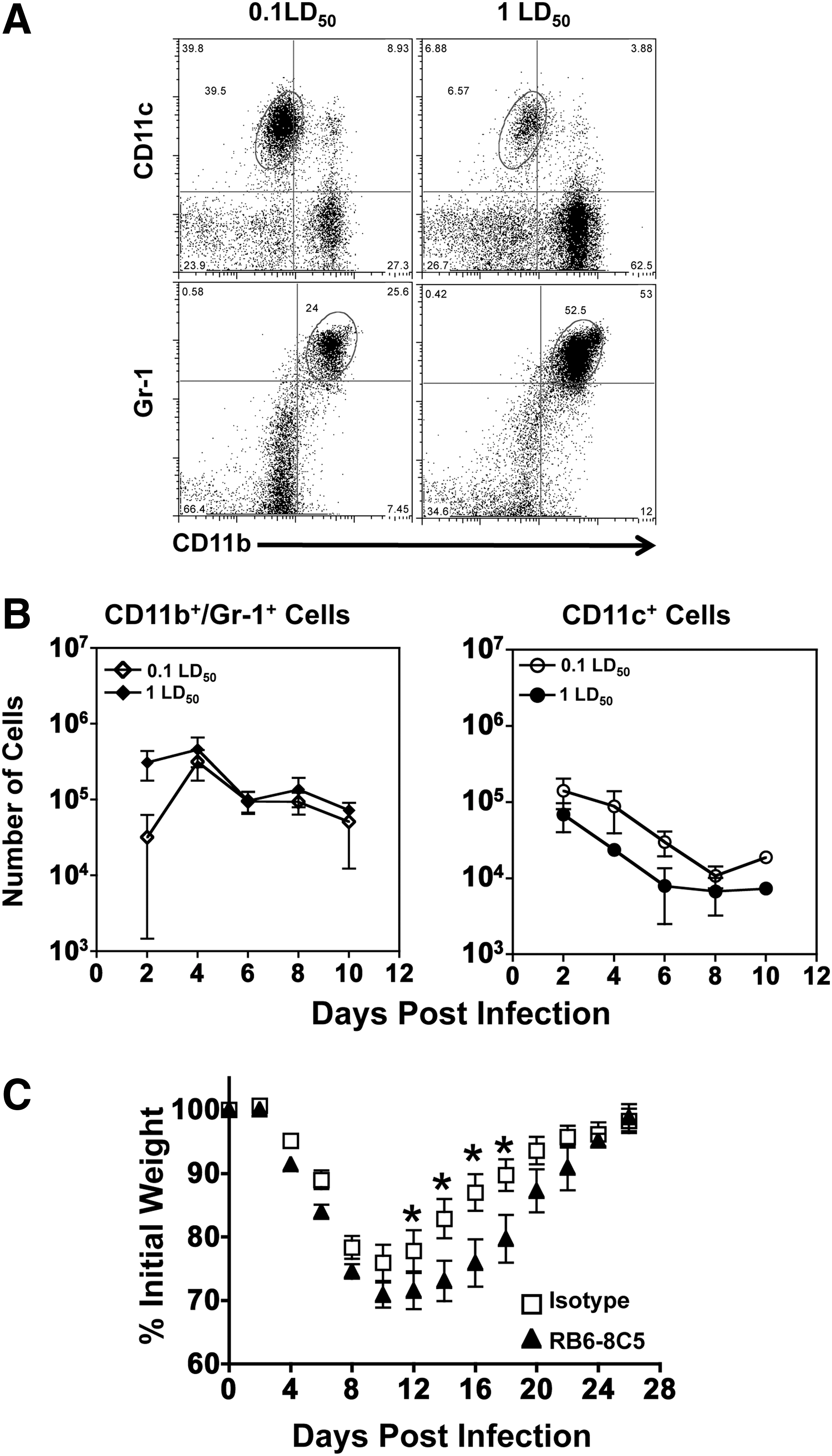
Blocking neutrophils enhances morbidity following influenza infection. Mice were infected intranasally with either a 0.1 LD50 or a 1 LD50 dose of IAV. BAL fluid was collected at each time point, and immune cells were stained and enumerated by flow cytometry.
T-cell migration but not CD4 function is similar in mice infected with different doses of influenza
Since blocking neutrophils did not enhance survival, we next hypothesized that early differences in innate immune cell influx and cytokine secretion affected the T-cell response. Surprisingly, no differences were observed in the number of infiltrating CD4+ or CD8+ cells in the BAL (Fig. 5A). NK cells showed differential kinetics between the two doses, with the peak NK influx at day 6 in the sublethal dose and day 4 in the lethal dose. The NK cell influx at the peak of the response was not significantly different (Fig. 5A) nor were GrB levels (data not shown). In contrast, CD4+ and CD8+ cell numbers continually increased in the airways over time and maintained nearly identical cell numbers between doses. To determine if there was a difference in effector function between the two doses, we examined Granzyme B (GrB) and peptide specific IFN-γ+ responses in both CD4+ and CD8+ cells. GrB expression was reduced in the lethal group, with significant differences in GrB MFI identified in CD4+ cells, suggesting that their cytolytic capacity may be impaired (Fig. 5B). IFN-γ+ secretion was also reduced in the CD4+ cell population in both the percentage (Fig. 5C) and total number of cells (Fig. 5D) at the site of infection. Interestingly, CD8+ responses were not impaired in any parameter we measured.
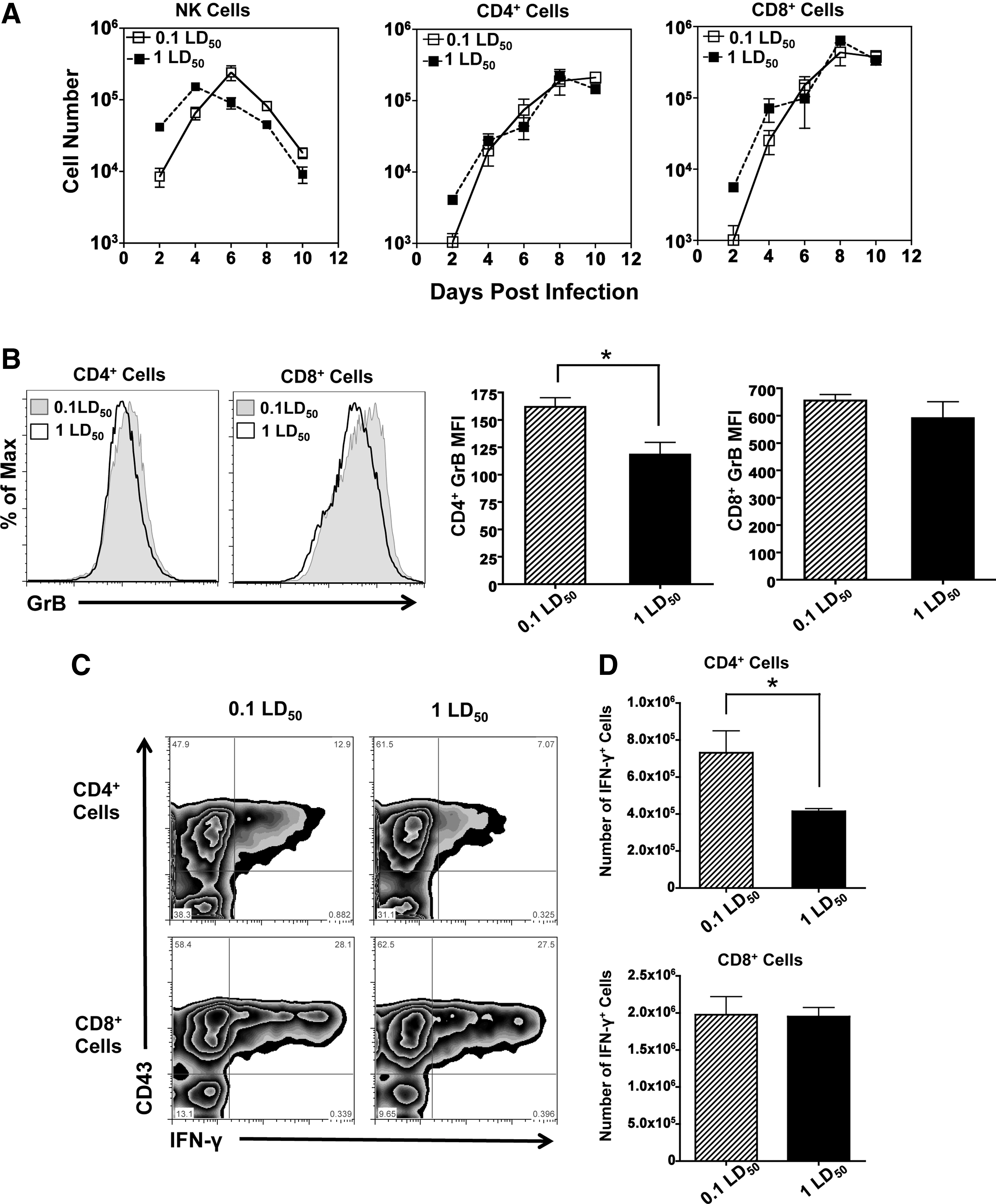
T-cell migration, but not CD4 function, is similar in mice infected with different doses of influenza.
Limiting viral replication enhances survival in lethally infected mice
Since early differences in cytokine production led to only modest differences in innate and adaptive immunity, we decided to examine lung pathology associated with the two viral doses. To ensure that all of the mice in the lethal dose cohort would succumb to infection, mice were infected with 2 LD50 of PR8. At both day 2 and day 6, infection with a high dose of IAV resulted in significantly increased pathology scores (Fig. 6A). When we examined the lung samples, infection with a lethal dose of IAV resulted in inflammation and infiltration of leukocytes within the bronchioles at day 2 (Fig. 6B). By day 6, infection with a high dose caused substantial inflammation (black arrowheads), massive infiltration of leukocytes as well as substantial epithelial cell hyperplasia (white arrowheads) and necrosis (black arrows).
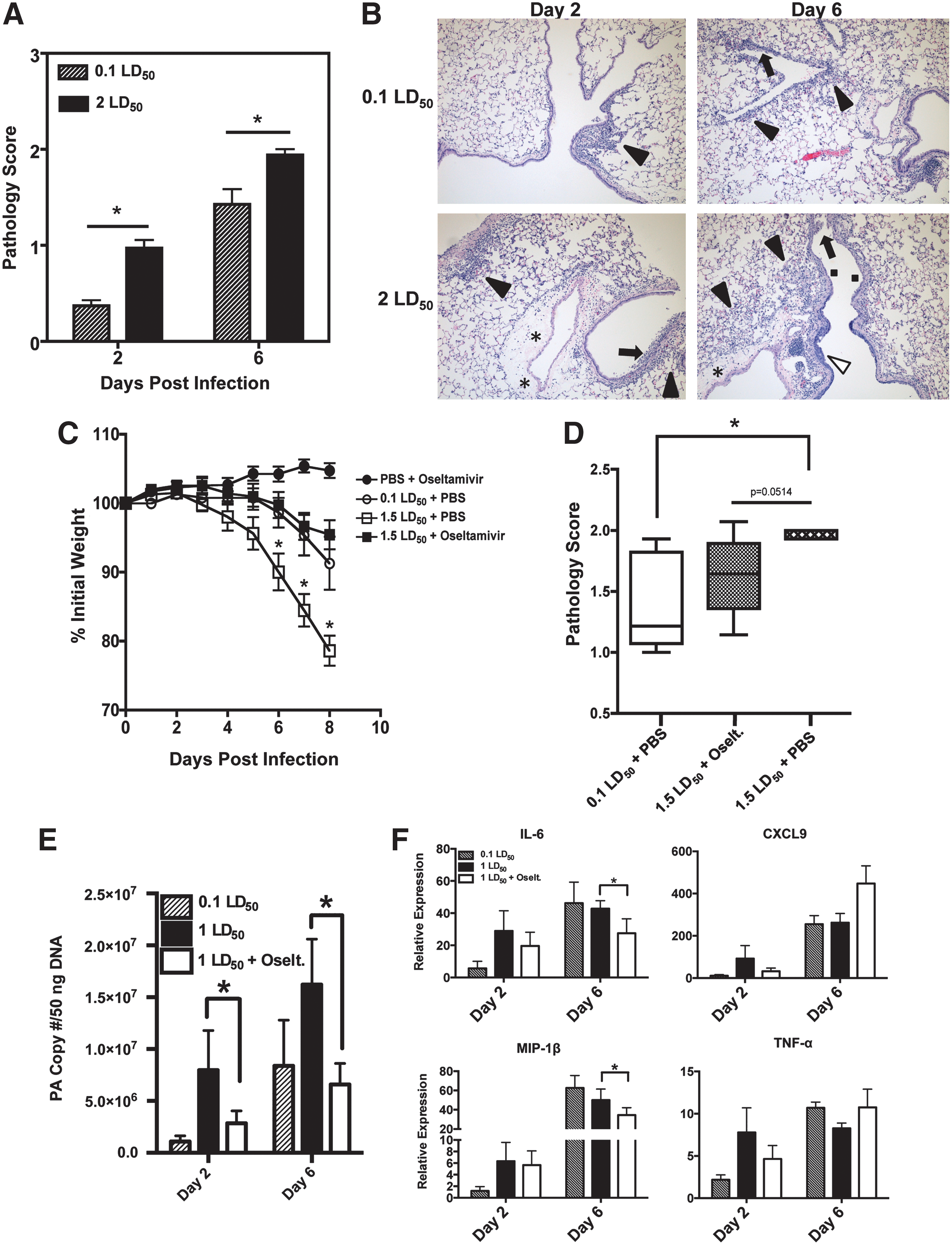
Limiting viral replication enhances survival in lethally infected mice.
To determine if extensive viral replication was the cause of mortality, the anti-influenza drug oseltamivir was used. Oseltamivir is a competitive inhibitor of the viral neuraminidase that prevents efficient replication by inhibiting viral release from infected epithelial cells. Following a lethal infection, mice given oseltamivir by oral gavage lost significantly less weight compared to mice given PBS (Fig. 6C). Furthermore, oseltamivir-treated mice showed nearly identical weight-loss kinetics to that of mice infected with 0.1 LD50. When lung samples were histologically examined at day 8, we saw that treatment with oseltamivir reduced the pathology in the lung compared to a 1 LD50 infection (Fig. 6D). In agreement with these findings, viral titers were also reduced in oseltamivir-treated mice. At day 2, significant differences in viral titers were observed between all groups, with lower titers from oseltamivir-treated mice compared to mice infected with a lethal dose. As expected, infection with 1 LD50 resulted in a statistically significant increase in virus at day 6. However, no differences in viral titer were observed between the 0.1LD50 group and mice treated with oseltamivir (Fig. 6E). Lastly, to determine if there was a relationship between inflammatory cytokines and oseltamivir treatment, qRT-PCR was performed in the lungs after infection. At day 2, the cytokine profiles mirrored the trends observed in the viral titer data, with a 1 LD50 infection generating the most robust responses, while a 0.1 LD50 infection induced the lowest levels (Fig. 6F). Mice given 1 LD50 and oseltamivir showed reduced levels of cytokines. However, these levels did not reach statistical significance. At day 6, levels of IL-6 and MIP-1β were significantly decreased in mice treated with oseltamivir, yet no differences were observed in CXCL9 or TNF-α expression. These data indicate that mortality following infection with a high viral dose may be not only the product of robust cytokine production, but also a consequence of high viral titers and efficient, unrestricted viral replication.
Discussion
Dissecting the contributions of IAV-mediated lung pathology over the course of an acute infection have proven difficult, as IAV infection is a complicated and dynamic event. In comparing two infectious doses, we sought to identify host-associated factors implicated in the mortality observed in mice after a lethal infection. Using this approach, we have outlined the kinetics of a sublethal and lethal IAV response, tracking multiple immune parameters over a course of 10 days. Infection with 1 LD50 was found to increase early cytokine production, viral replication, and pulmonary pathology manifested by onset of respiratory failure and mortality. These results have implications for clinical therapies; in particular, they highlight that therapeutics for influenza infection should target two processes—limiting both viral replication and exuberant cytokine responses in the lung microenvironment.
A number of findings in our study point to an early cytokine storm and immunopathology as the cause of mortality in lethally infected mice. First, cytokine dysregulation was observed following a 1 LD50 infection, with a number of cytokines elevated at day 2 postinfection. Recently, a clinical trial in humans found similar results with increased cytokine levels following severe influenza infection at early time points that were associated with increased disease (37). Second, knocking out single cytokines (28,34,39), their receptors (namely IL-6R; data not shown (35)), or neutrophils (Fig. 4C; (35)) enhances mortality, demonstrating that many cytokines, and even certain cell populations, have overlapping functions. The lack of any one cytokine implicated in IAV-mediated disease highlights the redundancy in the response and provides further evidence against the plausibility of knocking out a specific cytokine for therapeutic benefits. Third, treatment with oseltamivir not only rescued the lethal phenotype, but it abrogated lung pathology, making the lethal dose indistinguishable from a sublethal dose.
Our data show a differential innate cellular influx into the airways at early time points following infection. Infection with a lethal dose generates a high frequency of Gr-1+/CD11b+ cells and is a reflection of the massive influx of neutrophils into the lung and surrounding airways. Histological studies obtained from both early and late time points following IAV infection show marked and excessive accumulation of neutrophils in both the BAL and lung parenchyma (Fig. 6B). Despite these seemingly paradoxical observations, we and others have shown that neutrophil infiltration is required for survival (24,35) and can even enhance virus clearance (1,46). Besides their well-described antimicrobial roles, neutrophils also contribute to the healing process of infection by inhibiting their own accumulation, assisting in viral clearance, and secreting lipids that help macrophages initiate the repair process (25). Both the antiviral and homeostatic activity of neutrophils highlights the importance of this cell type in the IAV response.
In addition to differences in neutrophil trafficking, we also observed significant differences in IL-6 production in lethally infected mice at days 2 and 6 (Fig. 3). IL-6 potently modulates immune responses and has protective functions. Yet, excessive production in the lungs may compromise immunity and tip the balance toward pathology. At appropriate concentrations, IL-6 can act as a regulator of tissue inflammation by orchestrating immune cell apoptosis, chemokine production, and neutrophil influx. However, it can also contribute to pathogenesis by inducing the suppression of Treg cells, promoting excessive neutrophil/macrophage recruitment, as well as subsequent tissue destruction through protease activation (33). Thus, we conclude that the timing, duration, and amount of IL-6 are all important factors for generating a protective anti-influenza response.
Despite more lung damage and higher pathology scores, it was interesting to note that the T-cell influx into BAL (Fig. 5A) and the lung (data not shown) were virtually equivalent. Furthermore, the effector function of the CD8+ cells in the lung was not impaired when we examined intracellular levels of GrB or IFN-γ. Other experiments have shown that TNF-α may play a role in lung pathology mediated by CD8+ cells (26). Yet, in our system, TNF-α is equivalent between the two doses at days 6–10 when the adaptive response is generated (Fig. 3). Therefore, it may be the differences in the CD4+ cells that contribute to enhanced disease. Not only was there a decrease in IFN-γ secreting cells, a measure of Th1 effector function, but there was also a decrease in the GrB MFI. Given that CD4 CTL can protect against a lethal IAV infection in a perforin-dependent manner (14), these data further support the notion that CD4+ cells, via their cytotoxic mechanisms, help limit uncontrolled viral replication.
Taken together, the pathology we report after infection with a lethal dose of IAV may be due to a combination of a large inoculum of virus and its subsequent cytopathic effects on lung epithelial cells. Indeed, significant differences in viral titer were observed between the 1 LD50 and the mice treated with oseltamivir at day 6 (Fig. 6E) correlating with the pathology scores (Fig. 6D). However, one report suggests that viral titers are not the determining factor for disease progression, but a reduction in CD8 T-cell functionality, partially dependent on PD-1 expression (7). In our model, we did not observe a reduction in CD8 T-cell function as measured by IFN-γ and GrB. Our study was designed to examine the pathological effects of influenza using the same virus (PR8) with a different viral inoculum, while the study by Rutigliano et al. examined T-cell functions and cytokine production following infection with a high (PR8) or low (x31) pathological infection model. The differences in viral strains used could explain the differing results between these two models, again suggesting unidentified viral factors that impact the host immune response.
Previously, it has been demonstrated that IAV is detected by the immune system via TLR3 (16), TLR7 (4), and RIG-I (11,31). Interestingly, when we examined a panel of pattern recognition receptors, we saw that TLR9 transcripts were upregulated following infection (Fig. 2). This elevated expression could be due to the influx of NK cells, DCs, and macrophages that constitutively express high levels of TLR9. However, we might expect that other highly expressed PRRs on immune cells such as TLR3 and TLR7 would be increased to the same degree, yet we observed modest upregulation (two- to fourfold). This differential upregulation in lethally infected mice could be due to the high degree of pathology and necrosis observed in the lethal dose at day 2 (Fig. 6A). Because TLR9 is known to recognize unmethylated CpG motifs, we hypothesize that the high degree of epithelial cell death generates endogenous DAMP ligands. The breakdown and release of mitochondrial DNA that is characteristic of necrosis may explain the high TLR9 expression. Mitochondrial DNA has also been shown to activate neutrophils and induce degranulation in a TLR9-dependent manner (17), and in vivo administration of mitochondrial DNA has been shown to cause systemic lung inflammation (22). Though the issue of DAMP-mediated inflammation is still controversial, our data support this hypothesis from the high expression of TLR9 at early time points that correlate well with enhanced pathology scores. The mitochondrial DNA, which contains unmethylated CpG motifs (36,42), from epithelial cells could contribute to an inflammatory feed forward loop enhanced by the presence of DAMPs released into the lung microenvironment.
Other factors may also contribute to lethality following a high viral inoculum, including wound repair mechanisms and the restoration of lung homeostasis. Following a severe IAV infection, regeneration of epithelial cells and airway integrity may be impaired as well as the maturation of local lung resident stem cells (43). Additionally, the excess of anti-inflammatory molecules could inhibit sentinel immune cells leading to the development of bacterial superinfections and hyporesponsiveness to subsequent pathogenic insults. The pathways leading to wound repair and homeostasis following infection need to be further addressed, such as questions regarding the threshold of anti-inflammatory and repair responses following different viral inoculum. Many other factors need to be examined when determining the reasons for mortality in IAV-infected mice. Overall, the results of this study suggest that early viral replication is associated with increased pathology, inhibited CD4+ T-cell effector responses, and eventual mortality.
Footnotes
Acknowledgments
This work was supported by Public Health Service grants from the National Center for Research Resources (P30RR031151-03), the National Institute of General Medical Sciences (P30GM103509-03), and National Institute of Immunology, Allergy and Infectious Disease (R21-AI090438). AJV was supported by the Nebraska INBRE program (P20 RR016469). We thank Tyler Moore and Aspen Workman for the critical reading of this manuscript.
Author Disclosure Statement
No competing financial interests exist.