
Abstract
Sphingosine analogs display diverse immunoregulatory activities with curative potential in autoimmune diseases and viral infections. Recently, the sphingosine analog AAL-R was shown to increase DC activation upon TLR7 stimulation. Here, we investigated the effect of AAL-R on activation of dendritic cells (DCs) infected by lymphocytic choriomeningitis virus (LCMV). Concomitant treatment of LCMV-infected DCs with AAL-R enhanced DC maturation and DC ability to stimulate and expand antiviral CD8+ T cells. Importantly, AAL-R's stimulatory activity was abrogated in type I interferon (IFN) receptor-deficient DCs following LCMV infection. In support of this observation, AAL-R increased type I IFN production from DCs infected with LCMV. Taken together, the sphingosine analog could directly act on DCs to promote defensive host DC responses to the viral invasion via type I IFN signaling.
Introduction
S
Following virus infection, DCs are the most effective in presenting foreign antigens to stimulate the antigen-reactive T cells to fight against infection (2,24). Accordingly, many viruses seemed to have developed strategies to escape or suppress DC responses to prolong viral persistence in hosts (1,3,20,22). Therefore, identifying molecular mechanisms and ascertaining means to reverse viral suppression of DCs could provide an effective strategy to control virus infection.
Materials and Methods
Mice
C57BL/6, C57BL/6-Thy1.1+DbGP33–41 T cell receptor (TCR) transgenic (tg), and C57BL/6-IFNAR knock-out (ko) mice were used. Mice were bred and maintained in a closed breeding facility according to institutional guidelines. The animal protocol was approved by the Animal Care and Use Committee of University of Missouri-Columbia.
Dendritic cells
DCs were derived from bone marrow cells by culture with RPMI medium containing 20 ng/mL of recombinant mouse GM-CSF (PeproTech) for 10 days (29). DCs were infected with LCMV and then treated with AAL-R (1 μM) or its solvent (water) unless indicated differently. DC maturation was assessed at 1 or 2 days post-infection (dpi) (Fig. 1).
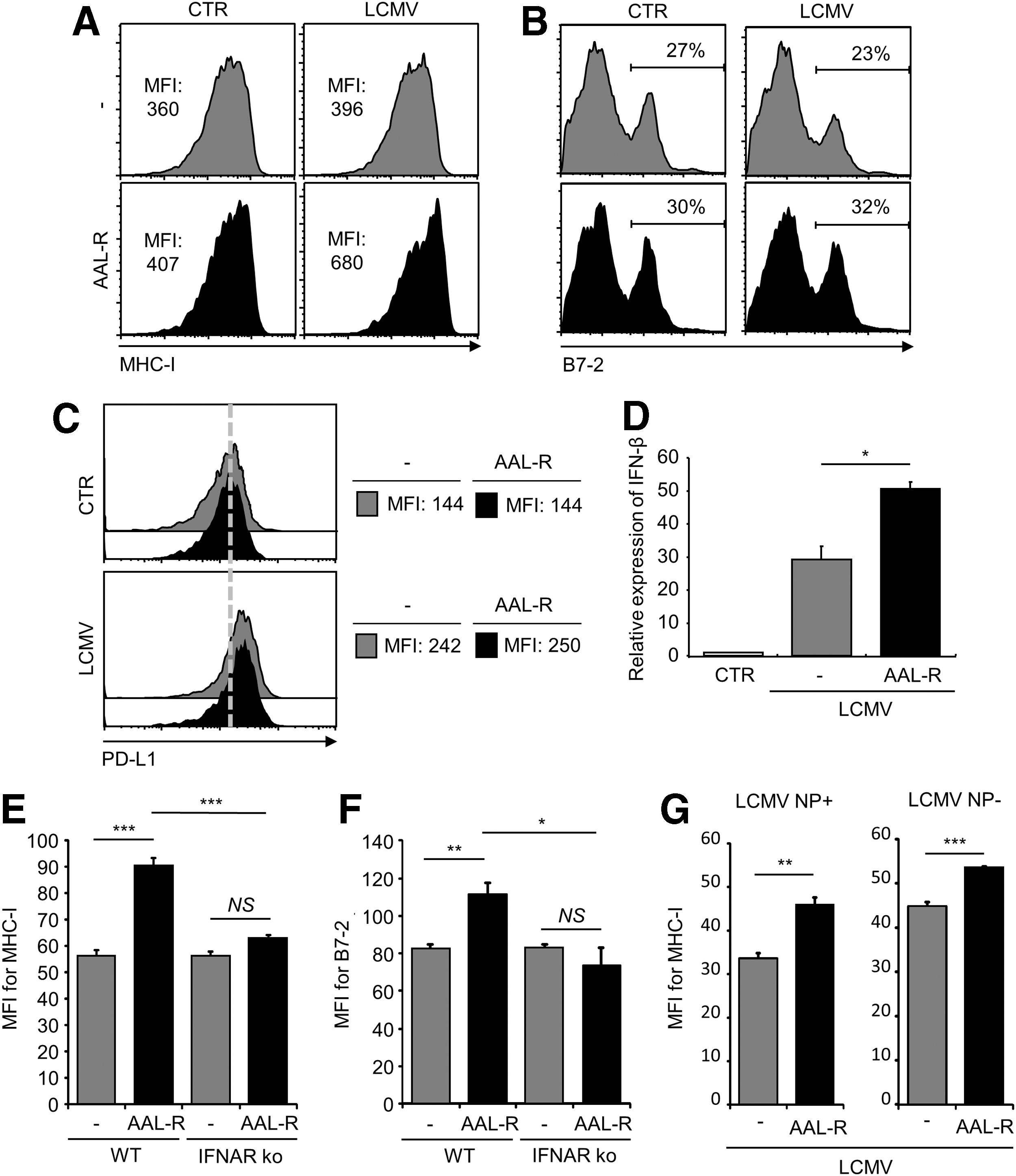
AAL-R enhances the maturity of LCMV-infected DCs via type I IFN signaling. Wt DCs were uninfected (control, CTR) or infected with LCMV Cl 13 (LCMV) and then treated with AAL-R (1 μM) or its solvent (−).
In vitro T cell proliferation assay
LCMV epitope GP33-specific CD8+ T cells were purified from LCMV epitope GP33-specific TCR tg mice by EasySep (Stemcell Technologies) and stained with CFSE (Invitrogen). Wt or IFNAR-deficient DCs were infected with LCMV Cl 13 at a multiplicity of infection (MOI) of 5 for 3 days, and then left untreated or treated with AAL-R for 2 or 6 h. GP33-specific CD8+ T cells were mixed with DCs at a DCs:T cells ratio of 1:10 (Fig. 2) or 1:30 (data not shown). After 3 days, the proliferation of antigen-specific T cells was evaluated by the decrease in CFSE fluorescence by flow cytometric analyses. The experiment was repeated three times with similar results.
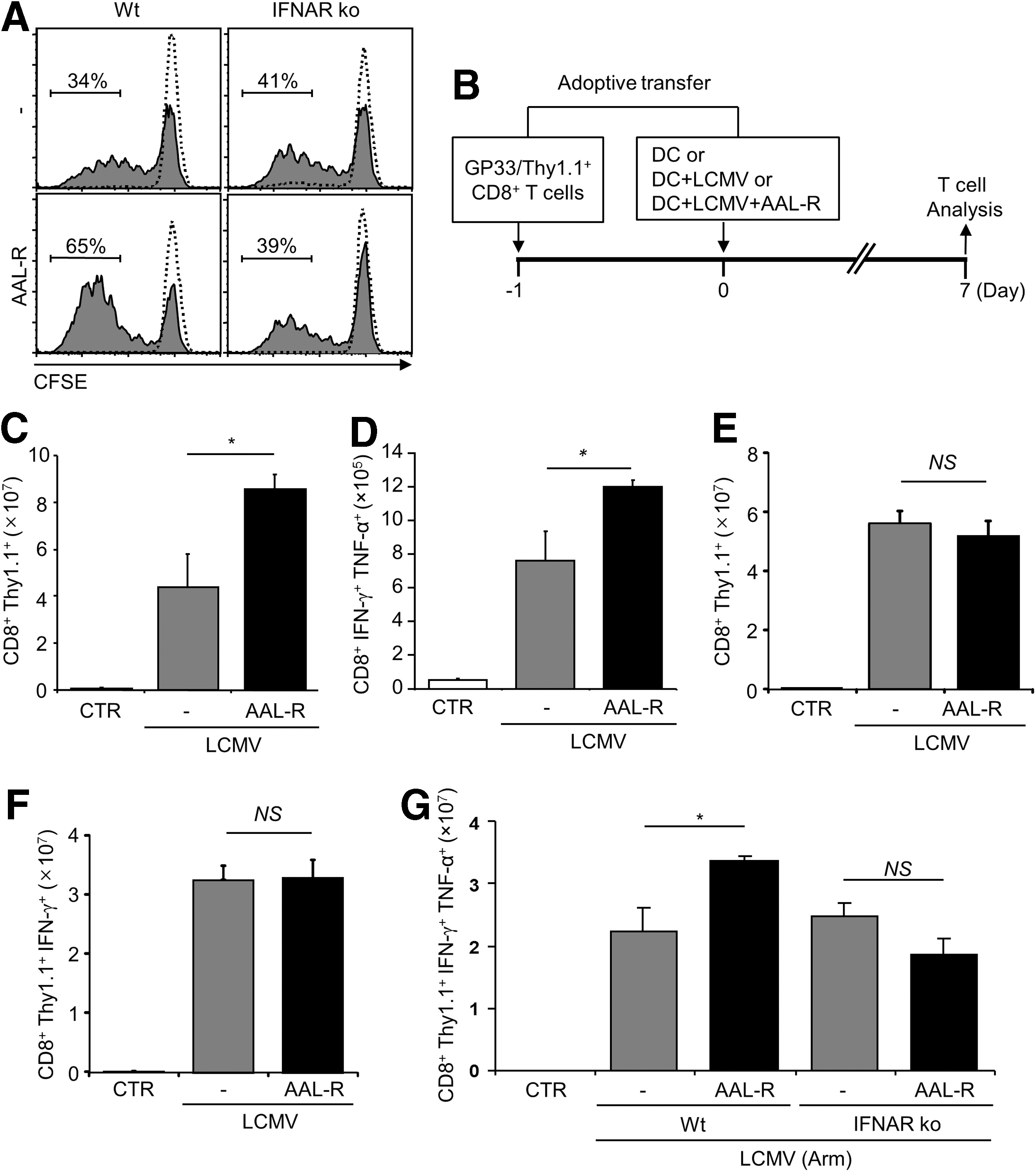
AAL-R enhances the function of DCs in promoting LCMV-specific CD8+ T cell activation via type I IFN signaling.
Flow cytometric analysis
Antibodies used in this study were specific for murine CD11c, MHC-I (H-2Kb), CD80 (B7-1), CD86 (B7-2), PD-L1, CD8α, CD90.1 (Thy1.1), TNF-α, and IFN-γ (BD PharMingen or e-Bioscience). LCMV-infected cells were detected by using a rat anti-LCMV nucleoprotein monoclonal antibody (VL-4 clone, BioXcell) (13). For the intracellular cytokine staining, cells were stimulated with LCMV-derived GP33–41 peptide (1.5 μg/mL) and treated with 5 μg/mL of brefeldin A (Sigma) for 6 h. Cells were then stained with antibodies and analyzed as described (5).
Real-time PCR
The cellular RNAs were isolated by TRI-reagent (Sigma-Aldrich) treatment followed by treatment with DNase I (Fermentas). After reverse transcription of RNAs, cDNAs were analyzed by real-time quantitative PCR (qPCR) for IFN-β expression. Primers for IFN-β (5′- CTC CAG CTC CAA GAA AGG ACG-3′, 5′- ATC TCT TGG ATG GCA AAG GCA -3′) and GAPDH (5′-TCA CCA CCA TGG AGA AGG-3′, 5′-GAT AAG CAG TTG GTG GTG CA-3′) were used. The qPCR was conducted with SYBR Green I chemistry on the ABI 7900 HT real-time PCR instrument. IFN-β cDNA quantities were normalized to GAPDH cDNA quantities measured in the same samples.
DC transfer assay
DCs were infected with LCMV at 5 MOI and left untreated or treated with AAL-R (1 μM) for 6 h. After washing three times, cells [3×105 (Fig. 2C) or 1×105 (Fig. 2E, 2F, and 2G)] were adoptively transferred intravenously into C57BL/6 Wt mice that received 3×105 (Fig. 2C) or 1×105 (Fig. 2E, 2F, and 2G) GP33-specific CD8+ T cells (Thy1.1+/GP33/CD8+ T cells) 1 day earlier. After 7 days, mice were euthanized for the analysis of virus-specific CD8+ T cells in spleens.
Statistical analysis
All error bars represent mean±standard error of the mean (SEM), and averages were compared using a bidirectional, unpaired Student's t-test.
Results and Discussion
Although the sphingosine analog AAL-R was shown to enhance TLR2, 7, or 9-mediated DC maturation (19), it is unclear if AAL-R exhibits immune stimulatory activity on DCs and DC-mediated T cell responses upon virus infection. Since TLR activations were shown to be involved in the host immune responses against LCMV infection (7,29,30), we evaluated the influences of AAL-R on LCMV infection-mediated DC responses. Infection with the immune suppressive Clone 13 (Cl 13) strain of LCMV is known to prevent efficient DC maturation (15,21,22,27). In agreement with it, LCMV Cl 13 infection did not upregulate MHC-I molecules (Fig. 1A) and co-stimulatory molecules B7-1 (data not shown) and B7-2 (Fig. 1B). However, treatment with AAL-R increased the expression level of MHC-I (mean fluorescence intensity, MFI: 396→680, Fig. 1A), B7-1 (data not shown), and B7-2 (B7-2 positive: 23%→32%, Fig. 1B) on DCs upon LCMV infection, when compared to that of untreated DCs.
While LCMV infection increased the amount of inhibitory molecule PD-L1 on DCs, the level of PD-L1 was not altered by AAL-R treatment (Fig. 1C), indicating that AAL-R's stimulatory activity is not due to the suppression of PD-L1 expression. Also, AAL-R treatment increased MHC-I surface expression on DCs infected with the Armstrong parental strain of LCMV (data not shown). These findings demonstrate that AAL-R enhances DC maturation upon LCMV infection.
The type I IFNs are known as strong inducer of DC maturation upon the virus infection (18), and the immunostimulatory activity of type I IFN on DCs appears to be nullified upon LCMV Cl 13 infection (27). Therefore, the possible association of the sphingosine analog's mode of action with the type I IFN signaling was evaluated. AAL-R increased IFN-β expression in LCMV-infected DCs compared to LCMV-infected, untreated DCs (Fig. 1D). Therefore, increased type I IFN production may result in enhanced DC maturation. Indeed, AAL-R was unable to increase the maturation of type I IFN receptor (IFNAR)-deficient DCs that are infected by LCMV, revealed by the unchanged expression of MHC-I (Fig. 1E), B7-1 (data not shown), and B7-2 (Fig. 1F). Thus, AAL-R-induced DC maturation upon LCMV infection is mediated by type I IFN signaling. We also compared MHC-I expression on LCMV-infected and uninfected DC populations in the same culture after LCMV inoculation onto DCs. Approximately 60% DCs were LCMV nucleoprotein (NP)-positive DCs in the experimental condition. AAL-R increased MHC-I expression on both LCMV NP-expressing (+) and nonexpressing (−) DC populations upon LCMV infection (Fig. 1G), although the effect was more pronounced on NP (+) DCs. Since AAL-R hardly upregulated DC maturation without LCMV infection and AAL-R's stimulatory activities on LCMV-treated DCs require type I IFN signaling, AAL-R could promote type I IFN production from LCMV-infected DCs to enhance maturation of uninfected DCs.
Next, we investigated the functionality of AAL-R-conditioned DCs in stimulating virus-specific CD8+ T cells. DCs that were infected with LCMV and then exposed to AAL-R strongly increased the rate of LCMV GP33–41 (GP33)-specific CD8+ T cell proliferation, compared to untreated control DCs in vitro (34%→65%, Fig. 2A). The data indicate that AAL-R enhances the T cell stimulatory function of the virus-infected DCs. However, the increased T cell proliferation was not repeated when DCs were lacking type I IFN receptor (41%→39%, Fig. 2A), which is consistent with the result from DC maturation study shown in Figure 1.
A final series of experiments was conducted to assess the consequences of AAL-R-conditioning for DCs in vivo. AAL-R-treated, LCMV-infected DCs (AAL-R-DCs) or untreated, LCMV-infected DCs were adoptively transferred into adult C57BL/6 wild type (Wt) mice that received Thy-1-mismatched, LCMV epitope GP33-specific CD8+ T cells 1 day earlier. At 7 days post-transfer, the expansion of virus-specific, antiviral CD8+ T cells was assessed (Fig. 2B). LCMV-specific CD8+ T cells retrieved from AAL-R-DCs-recipient mice (Mean±SEM, 8.6±0.6×107) expanded approximately two-fold more than T cells stimulated by LCMV-infected, untreated DCs (4.4±1.4×107) (Fig. 2C). Similarly, the number of endogenous IFN-γ/TNF-α-expressing GP33-specific CD8 T cells in AAL-R-DCs-recipient mice were significantly higher than that in LCMV-infected, untreated DCs-recipient mice (Fig. 2D). In contrast, when IFNAR-deficient DCs were infected, treated with AAL-R, and then adoptively transferred into Wt mice, they failed to promote CD8+ T cell responses compared to untreated DCs (Fig. 2E and 2F). Similar results were recapitulated when DCs were infected with LCMV Armstrong (Arm) instead of Cl 13 strain (Fig. 2G). Collectively, we conclude that the sphingosine analog AAL-R induces maturation of LCMV-infected DCs and elevates the capacity of DCs to stimulate virus-specific T cells via type I IFN signaling.
The sphingosine analog was known to decrease cytokine production from lung endothelial cells upon influenza virus infection in vivo (26). However, AAL-R displayed immune stimulatory activity on DCs that were stimulated with TLR7 agonist and enhanced type I IFN production (19). In this study, we found that AAL-R exhibits immune stimulatory activity on DCs even when the cells were infected with an immune suppressive LCMV strain with enhanced production of type I IFN. Therefore, the differential immunomodulatory activity of the analog could depend on several factors including cell types and the nature of pathogens.
The therapeutic application of FTY720 was attempted to resolve persistent virus infection (8,28). However, systemic administration of FTY720 into mice did not alleviate LCMV Cl 13-induced T cell exhaustion and failed to clear virus infection (28). Since AAL-R was shown to enhance DC maturation and DC-mediated antiviral CD8 T cell responses when DCs were infected by LCMV, the sphingosine analog displays an immunostimulatory activity on LCMV-infected DCs. However, the sphingosine analog could act on diverse cell types when instilled in vivo, representing the complexity of sphingosine system (4,16). For instance, FTY720/AAL-R is known to modulate lymphocyte trafficking (12) and cytokine productions from endothelial cells (26), resulting in immune suppression. It is noteworthy that systemic administration of FTY720 following LCMV infection did not affect T cell induction, expansion, and memory development, although it hindered effector T cells' peripheral homing (14). Therefore, systemically instilled sphingosine analogs following LCMV infection act on immune cells including T cells to regulate cell trafficking and possibly regulate some other cellular functions to suppress host immune system, which may have masked or compromised the sphingosine analog's stimulatory effects on DCs in vivo.
Our findings suggest that the sphingosine analog could exhibit immune stimulatory activity especially in DCs upon virus infection. Therefore, regulation of sphingolipid signaling in DCs could be utilized for the development of DC-based therapeutic vaccine approach to the resolution of virus infections.
Footnotes
Acknowledgments
This work was supported by NIH/NIAID Grants R21AI088363 and R01AI091797 (B.H). We thank Michael Oldstone and Hugh Rosen (The Scripps Research Institute) for their kind provision of C57BL/6-Thy1.1+DbGP33–41-TCR tg and C57BL/6-IFNAR ko mice, and AAL-R, respectively. Also, we thank facilities of Cell & Immunology Core and Animal Care at MU.
Author Disclosure Statement
No competing financial interests exist.