
Abstract
The VP6, the group antigenic rotavirus (RV), is highly conserved and the most abundant, constituting about 39% of the viral structure proteins by weight. The high degree of identity (>87%–99%) in the primary amino acid sequences suggests VP6-based vaccines could potentially provide heterotypic protection. Although some efforts have been made toward producing recombinant rotavirus VP6 vaccines, the native VP6 is still unsatisfactory as an optimal vaccine. The major neutralizing antigenic epitopes that exist on VP4 or VP7 are not on the native VP6, and as a vector the native VP6 lacks insertion sites that can be used for insertion of foreign epitopes. In this study, a new foreign epitope presenting system using VP6 as a vector (VP6F) was constructed on the outer surface of the vector six sites that could be used for insertion of the foreign epitopes created. Using this system, three VP6-based VP4 epitope chimeric proteins were constructed. Results showed that these chimeric proteins reacted with anti-VP6 and -VP4 antibodies, and elicited antibodies against VP6 and VP4 in guinea pigs. Antibodies against VP6F or antibodies against the chimeric proteins neutralized RV Wa and SA11 infection in vitro. It is optimistic that the limitation for using the native VP6 as a vaccine candidate or vector will be solved with our proposed approach. It is expected that this VP6-based epitope presenting system and the VP6-based VP4 epitope chimeric proteins will be valuable for and contribute to the development of novel RV vaccines and vaccine vectors.
Introduction
G
There are two oral live RV vaccines are currently available, Rotarix, a monovalent vaccine derived from a clinical human isolate of serotype G1P1A[8], and Rotateq, a pentavalent vaccine containing human RV (HRV) VP7 proteins of serotypes G1, G2, G3, and G4 and genotype P1A[8] on a G6P[5] bovine RV background (WC3 strain) (35,41). Clinical trials of the current RV vaccines reported at least 85% protection rates against severe diarrhea (35,41,42). However, a risk of adverse effects has been associated with some live vaccines. Among these, intussusception was associated with a previously licensed RV vaccine (29). A small increased risk of intussusception has been reported in association with the current licensed RV vaccines after the first dose in some countries (2,3,31,40). Other data raise concerns regarding the safety of live RV vaccines in severely immunocompromised patients (31). Furthermore, some live vaccines may carry risks of the presence of adventitious agents in the vaccine (16,37). Because live rotavirus vaccines are found to have intrinsic limitations, it is necessary to develop vaccines that are more efficacious in developing countries.
As an alternative to live vaccines, virus-like particles (VLPs) have been evaluated as recombinant nonreplicating candidate vaccines. The VLPs self-assemble from viral capsid proteins, and resemble native virus structurally and antigenically (7). They are noninfectious due to lacking of viral nucleic acids. However, as live vaccines, wide protection from heterogeneous genotypes infection must be addressed.
The VP6 is highly conserved and the most abundant, constituting approximately 39% of the viral structure proteins by weight. A few candidate VP6-based vaccines have been studied since antibodies against VP6 are highly cross-reactive among all RVAs; therefore, VP6-based vaccines could potentially provide heterotypic protection (32). Anti-VP6 IgA has protective efficacy in vivo by inhibiting viral transcription at the start of the intracellular phase of the viral replication (12). In addition, immunization with VP6 may prime the immune system for enhanced production of neutralizing antibodies against the external proteins (VP7 and VP4) upon challenge with homotypic or heterotypic viruses (9). Anti-VP6 antibodies have a neutralizing activity against rotavirus VP6 proteins in vitro; this was confirmed by previous experiments that demonstrated anti-VP6 Llama-derived single-chain antibody fragments (VHH) had neutralizing activity against VP6 in vitro. VP6 was reported to be involved in RV cell entry via its binding to the cellular heat shock protein (hsp70) (15); it might be related to the presence of neutralizing epitopes in VP6. Even a short fragment of VP6 could provide significant reduction in virus infectivity in vitro (8).
Recombinant VP6 (rVP6) and double-layered (dl) 2/6-virus-like particles (VLPs) were considered as the simplest RV subunit vaccine (1,20). Both the rVP6 and dl2/6-VLPs induced a balanced Th1-type and Th2-type response and high levels of serum IgG antibodies with cross-reactivities against different RV strains (Wa, SC2, BrB, 69M, L26, WC3, and RRV).
Although some progress has been achieved, it is still uncertain to use the native VP6 as an optimal vaccine or vector. First, native VP6 lacks neutralizing antigenic contents of the VP4 or VP7 as the major antigenic protein, resulting in unsatisfactory immunogenicity. Second, the native VP6 as a vector lacks proper insertion sites that can be readily used for insertion of foreign epitopes. Therefore, the native VP6 needs to be modified so that it can be practically used as a vector. Furthermore, for development of VP6-based vaccines, the epitopes derived from the VP4 or VP7 should be included.
The VP4 is a main protective antigen that induces neutralizing antibodies. The VP4 is a nonglycosylated protein, containing serotype-specific sites between aa80–aa180. The VP4 is the major crossing-neutralizing antigen, has characteristics of hemagglutinin and trypsin cleavage enhancing virus infectivity (10). With only a single serotype-specific VP4 protein, neither a live attenuated vaccine nor recombinant vaccine is able to fully protect from heterogeneous RV infections. In theory, as the group (subgroup) antigen with high identity and the characteristics mentioned above, the VP6 carrying epitopes with high homology derived from the VP4 should be considered as remedy to this defect.
Some epitopes had been described in previous studies. Six peptides on the VP4 (residues aa1-10, aa35-44, aa55-66, and aa223-234, aa296-313, aa381-401) that contained sequential antigenic determinants were cross-reacting neutralization epitopes (18,19,38). These findings indicated that these sequential epitopes may also be important for the RV recombinant epitope chimeric vaccines.
In the present study, a foreign epitope presenting system using VP6 as a vector (VP6F) was created and, three VP4 epitope chimeric recombinant vaccines constructed based on the VP6F vector system, and their immunoreactivities were characterized. It is hopeful that the limitation for using of the native VP6 as an optimal vaccine or vector will be solved with our proposed approach.
Materials and Methods
Molecular structure determination of the VP6 protein of RV strain TB-Chen
Molecular structure of the VP6 protein of RV strain TB-Chen (RVA/Human-wt/CHN/TB-Chen/1996/G2P[4] (6,27) was determined as described below. Briefly, with protein blast software supplied in the NCBI (NCBI (
Construction of a vector for displaying foreign epitopes based on the VP6 protein
Since the VP6 protein had a specific conformation (Fig. 1A), it was used as a vector for delivery of a variety of foreign epitopes from other infectious agents and endogeneous proteins. In order to improve the presentation and immunogenicity of foreign epitopes, in the present study, using molecular cloning procedures and genetic recombinant techniques (5), the VP6 coding cDNA was cloned from RV strain TB-Chen, and inserted six endonucleotide enzyme sites, Sac I, BspT104 I, Kpn I, Bln I, Sac II and Xho I, in the six regions, creating a new recombinant gene that codes for a recombinant protein (VP6F) with six foreign epitopes insertion sites designated I1, I2, I3, I4, I5, and I6, on its exposed surface. These insertion sites were designed at six corresponding loops in the naïve VP6 protein molecular structure. The molecular structure of the resultant vector protein VP6F was then determined as described above (Fig. 1B).
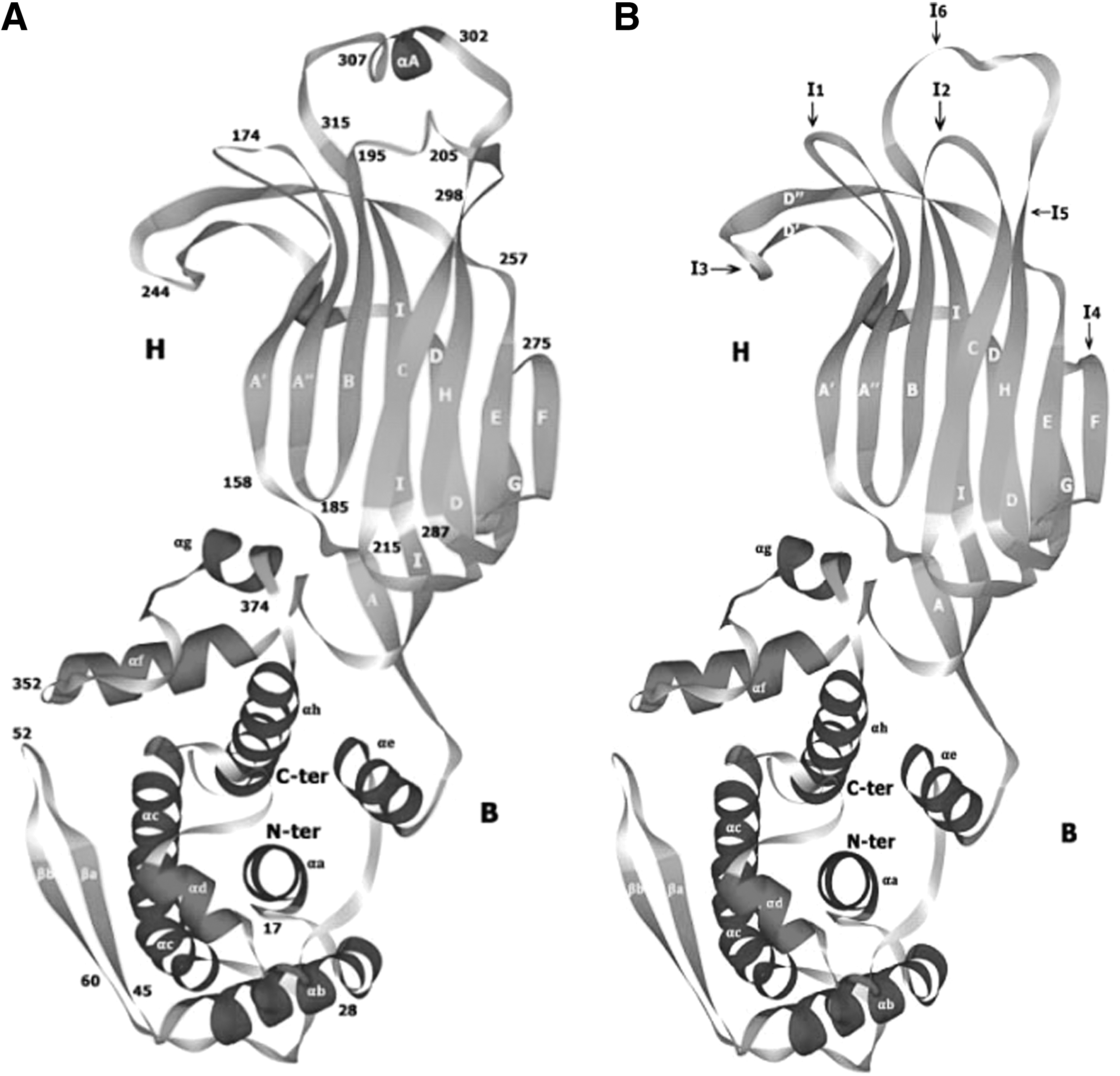
Three-dimensional molecular structure of the naïve VP6 protein
Epitopes, recombinants, and expression
Three epitopes derived from the VP4 of rotavirus were studied. Epitopes 174S/P[4]223, 197B/P[4]56, and 298S/P[8]/296, corresponding to the residues aa223-234 (LPPIQNTRNVVP), aa56-71 (NDSTTVEPVLDGPVQP), and aa296-313 (KAANYQYNYLRDGEQVTA), respectively, were selected to construct recombinant epitope chimeric vaccines.
Three pairs of specific oligonucleotides, 174S/P[4]223f (5′- C CCA CCA ATT CAA AAT ACT AGA AAT GTA GTT CCA GAG CT -3′)/ 174S/P[4]223r(5′- C TGG AAC TAC ATT TCT AGT ATT TTG AAT TGG TGG GAG CT-3′), 197B/P[4]56f (5′ -CG AAT GAT TCA ACT ACA GTG GAA CCA GTT TTA GAT GGT CCT TAT CAA CCT T-3′)/ 197B/P[4]56r (5′- CGA AGG TTG ATA AGG ACC ATC TAA AAC TGG TTC CAC TGT AGT TGA ATC ATT-3′), and 298S/P[8]/296f (5′- GG AAG GCA GCA AAT TAT CAA TAT AAT TAC TTA CGT GAC GGT GAA CAA GTA ACC GCA CCG C-3′)/ 298S/P[8]/296r (5′- GG TGC GGT TAC TTG TTC ACC GTC ACG TAA GTA ATT ATA TTG ATA ATT TGC TGC CTT CCG C-3′), representing the corresponding eiptopes were synthesized and inserted into the BspT104 I, Sac I, and Sac II cloning sites at the positions of I1 (174S/P[4]223), I2 (197B/P[4]56), and I5 (298S/P[8]/296-313) on the VP6F protein vector by means of genetic recombination techniques (5). The VP6F/VP4 epitope chimeric protein coding gene was put into the plasmid pETL that was derived from pET-3a, and four recombinant plasmids directing the expression of the chimeric proteins were constructed by genetic recombination techniques as previously described (5), pETP6F, pETP6F197B/P[4]56, pETP6F174S/P[4]223, pETP6F298S/P[8]296. pETP6F carried the naïve VP6 gene, pETP6F197B/P[4]56 carried the VP6F gene with epitope 197B/P[4]56 inserted in I2 site, pETP6F174S/P[4]223 carried the VP6F gene with epitope 174S/P[4]223 inserted in I1 site, and pETP6F298S/P[8]296 carried the VP6F gene with epitope 298S/P[8]296 inserted in I5 site.
The above recombinant plasmids were further sequenced to confirm that the epitope sequence was correctly inserted and transformed into E. coli BL21(DE3) cells and expressed in LB medium supplemented with 200 μg/mL ampicillin at 37°C. Cells were collected, lysed, and chimeric proteins were extracted and analyzed by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (10% SDS-PAGE) as previously described (5).
Protein purification and concentration determination
The expressed proteins were purified by SDS-PAGE as previously described (5). Briefly, the proteins were separated on 10% SDS-PAGE and dyed with 150 mmol/L KCl. According to the molecular weight of the protein, the band of gel containing only the interesting chimeric protein was cut off and put into a column filled with polyacrylamide gel (15%) on its bottom. Electrophoresis was performed at 20 mA, 4°C for 15 h, and the chimeric protein was enriched in the vial that was linked to the column bottom. The protein was further dialyzed in 10 mmol/L phosphate-buffered saline (PBS) (pH 7.4) for desalination and renaturation. The concentration of the expressed proteins was determined by Lowry's method (22).
Animals and immunization
Guinea pigs (5–7 weeks of age) used in this study were purchased from China Medical Primates Center, Kunming, China, and bred within our animal facility. All research performed on guinea pigs in this study complied with national and institutional guidelines set forth by the Institute of Medical Biology Animal Care and Use Ethic Committee. All studies were approved by the Institute of Medical Biology Animal Care and Use Ethic Committee.
All guinea pigs were screened for antibodies against RVA by neutralization test upon arrival to ensure absence of preexisting immunity from prior exposure or maternally-derived antibody. Recombinant vector protein rVP6F, chimeric protein that carrying VP4 epitope, P[4]223, P[4]56, or P[8]296, and reference rotavirus Wa, SA11 were used as immunogens. Four guinea pigs were used for each immunogen inoculation. For rVP6F, P[4]223, P[4]56, and P[8]296 inoculation, each animal was inoculated subcutaneously at the hinder leg with 120 μg in 100 μL solution (15 mM Tris, 150 mM NaCl, pH 7.0) in the absence of adjuvant. For Wa and SA11 inoculation, 1×108 TCID50 virus was used for each animal each time. At the same time, four guinea pigs were used as negative control and mock-inoculated with 10 mmol/L of PBS. Each animal had three inoculations at 2-week and then 1-week intervals. Guinea pigs were bled from the heart at the 5th day post the last inoculation and antibody levels were measured by Western blot and neutralization test.
Immunofluorescence analysis
The MA104 cell monolayers on the coverslip glass cultured in Minimum Essential Eagle Medium (MEM) were washed twice with PBS and infected with RV strains SA11 that were previously digested with acetylated trypsin (10 μg/mL). The coverslips were taken out at 12 h after infection, washed twice with PBS, fixed with prechilled methanol, and rehydrated for 10 min at 4°C with 70%, 30%, and 10% of prechilled ethanol, respectively. After washing with PBS, coverslips were incubated for 1 h at 37°C with antisera (1:400 diluted with 0.1% of bovine serum albumin) collected from guinea pigs inoculated with SA11, VP6F, chimeric protein 6F/P[4]223, 6F/P4[56, 6F/P[8]296, or with the serum derived from pre-immune guinea pigs or the guinea pigs mock inoculated with PBS, respectively. After washing, coverslips were incubated at 37°C for 1 h with FITC-labeled goat anti-guinea pig IgG (1:100 dilution), and fluorescence was detected under microscope. Meanwhile, as controls, fluorescence analysis in noninfected cells and SA11 infected cells with pre-inoculated sera or with negative sera (inoculated with PBS) were carried out.
Western blot analysis
Western blot was performed as previously described (5). Briefly, chimeric proteins (about 4 μg each) were separated on 10% SDS-PAGE, transferred onto PVDF (polyvinylidene fluoride) membrane and detected with antibodies in immunized guinea pig serum (at 1:400 dilution). Bound antibody was detected with goat anti-guinea pig immunoglobulin G (IgG) antibody conjugated to horseradish peroxidase (at 1:2000 dilution) and DAB (3,3′-diaminobenzidine tetrahydrochloride, Sigma).
Cell culture infection and determination of tissue culture infectious dose50 (TCID50)
MA104 cells were used for preparation and determination of RV infectious doses. Briefly, when cells grew in MEM supplemented with 10% calf serum to confluent monolayer, medium was removed and cells were washed twice with PBS, incubated for 1 h with acetylated trypsin (10 μg/mL, 37°C for 1 h) pre-treated RV strains SA11, with gentle rocking at 15-min intervals. The virus solution was removed and cells were then incubated in maintenance MEM medium (without calf serum) at 37°C/5% CO2. When cytopathic effect (CPE) appeared among ≧75% of cells, whole cells and supernatant were frozen and thawed three times. After centrifugation at 4000 rpm at 4°C for 30 min, supernatant was taken and virus infectious doses were determined by TCID50 using CPE with Kärber's method (16).
Neutralization test
The RV strains Wa and SA11 were used to estimate the neutralizing reactivity of antibodies elicited in serum of guinea pigs inoculated with the chimeric proteins. Briefly, 50 μL of acetylated trypsin virus solution containing 100 TCID50 was mixed with equal volume of the guinea pig antiserum at 2-fold serial dilutions, incubated for 1 h at 37°C. The mixture was then added onto MA104 cell monolayers in 96-well plates. Each dilution was determined with 4 wells. The cell plates were incubated in 5% CO2 incubator for 48 h and CPE was observed. Neutralizing titers were defined as the highest dilution of antiserum that protected 50% of cells from virus infection.
Results
Molecular structure of the naïve VP6 protein of TB-Chen and the VP6F protein as a vector for displaying foreign epitopes
The predicted molecular architecture of the native VP6 protein (397 aa) of TB-Chen was almost identical to that of RF strain (23). Briefly, the polypeptide chain of VP6 of TB-Chen is folded into two distinct domains, B and H, shown in the ribbon diagram (Fig. 1A). Domain B, at the base of the molecule, consists of a bundle of eight α-helices (αa–αh) derived from two segments of the polypeptide chain. The first segment, containing helices αa–αe, is formed by the 150 N-terminal amino acids. The second segment (residues 335–397 at the C-terminal end) contains the remaining three helices. Domain H (residues 151–334) was at the top of the molecule, folding into a β-sandwich. This domain is an insertion between consecutive helices αe and αf of domain B. The CHEF β-sheet lies in the outer exposed side of the molecule, the latter β-sheet is augmented in VP6 by β-hairpin A′A″, N-terminal to strand B. The connection between strands D and E forms another β-hairpin, D’D. At the top of H domain formed six loops that exposed outside, sited between β-hairpin A’A” (Loop 1), β-sheet BC (Loop 2), β-hairpin D’D” (Loop 3), FG (Loop 4), α-helices αA at the top of sheet H (Loop 5), and α-helices αA at the top of sheet I (Loop 6) (23).
By means of molecular cloning and genetic recombinant techniques, a recombinant gene that codes for a recombinant protein (VP6F) with six foreign epitopes insertion sites on the top of the six loops was created. The basic structural feature of the VP6F was almost identical to that of the naïve VP6 of TB-Chen (Fig. 1B). The molecular structure of VP6F as predicted showed that the bone structure of VP6 was fully maintained in VP6F, and the six foreign epitopes insertion sites designated I1, I2, I3, I4, I5, and I6, were as expected on its exposed surface (Fig. 1B).
Expression of chimeric proteins
The recombinant plasmids pETP6F, pETP6F174S/P[4]223, pETP6F197B/P[4]56, and pETP6F298S/P[8]296 were sequenced to confirm that the epitope sequences were correctly inserted and transformed into transformed into competent BL21(DE3) cells and expressed. The recombinant proteins were expressed in inclusion body form in the transformed cells. The expressed proteins, recombinant vector protein VP6F, chimeric proteins 6F/P[4]223 that carries the epitope 174S/P[4]223, 6F/P[4]56 that carries the epitope 197B/P[4]56, and 6F/P[8]296 that carries the epitope 298S/P[8]/296, had molecular weights of 43.2, 44.6, 45.0, 45.5, kDa, as expected, and possessed 46.7, 45.2, 47.0, 51.0% of the total proteins of the transformed cells, respectively (Fig. 2). The chimeric proteins were purified (Fig. 3) and used to immunize guinea pigs.
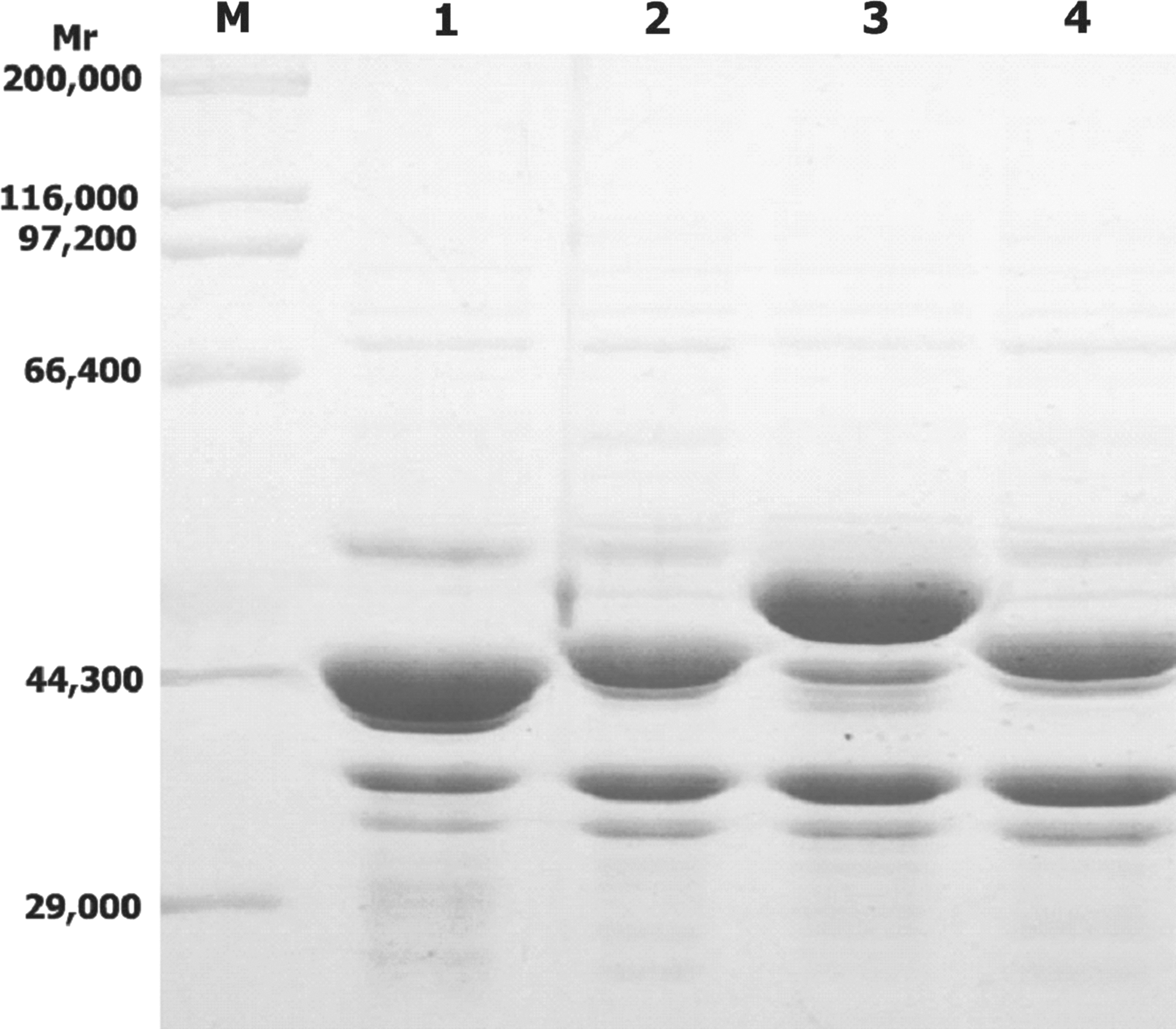

Immunoreactivity of the chimeric proteins
The immunological reactivity of the chimeric proteins was detected by Western blot. Western blot test results showed that all the chimeric proteins carrying the VP4 epitopes could be specifically recognized by antibodies in all the four sera derived from guinea pigs inoculated with recombinant VP6 (Fig. 4, only one result is shown), or VP4 protein (Fig. 5. only one result is shown). There was not any immunoreactivity of serum derived from pre-immune or negative control animals observed with VP6F or the chimeric proteins carrying VP4 epitopes (data not shown).
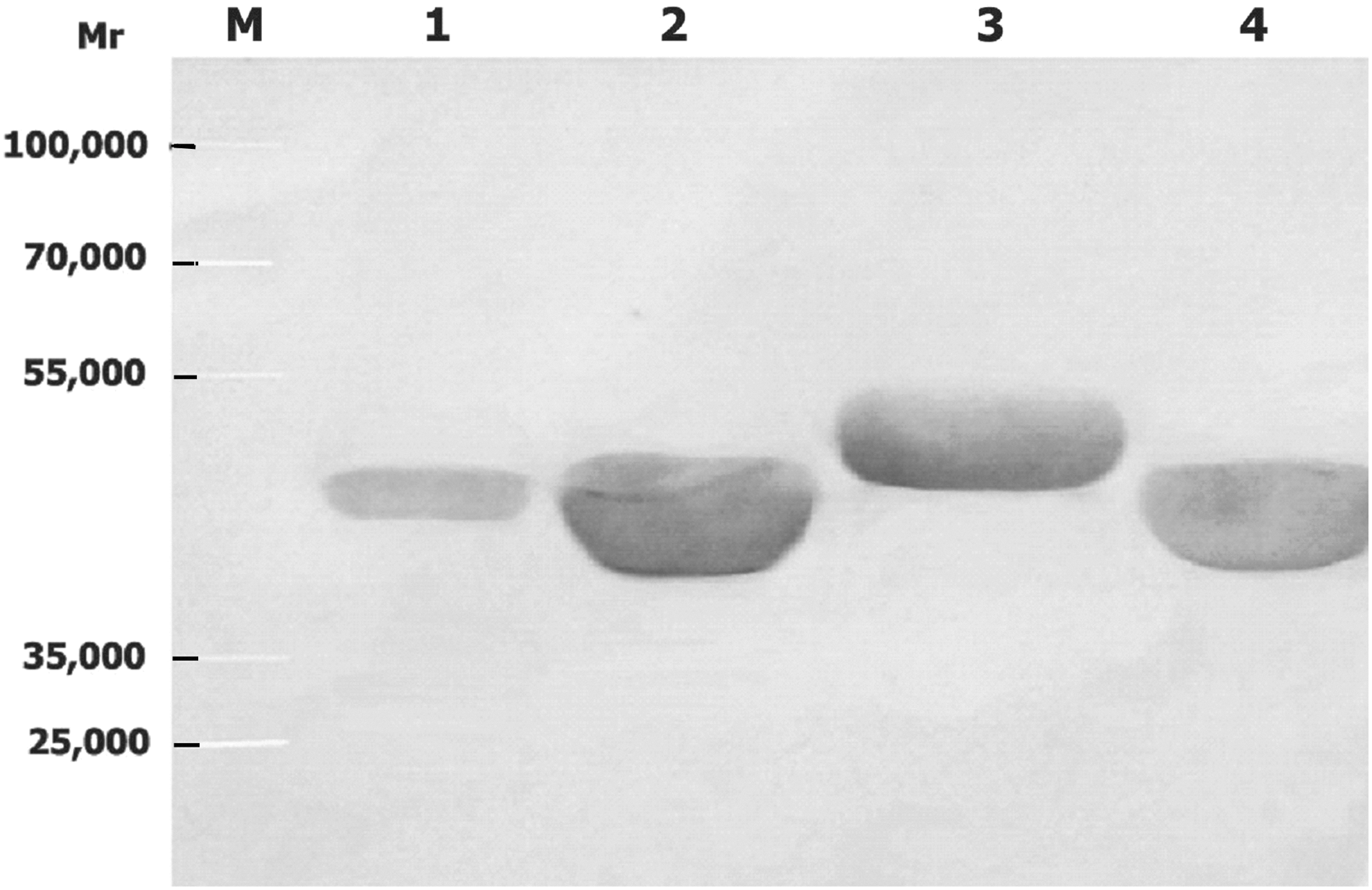
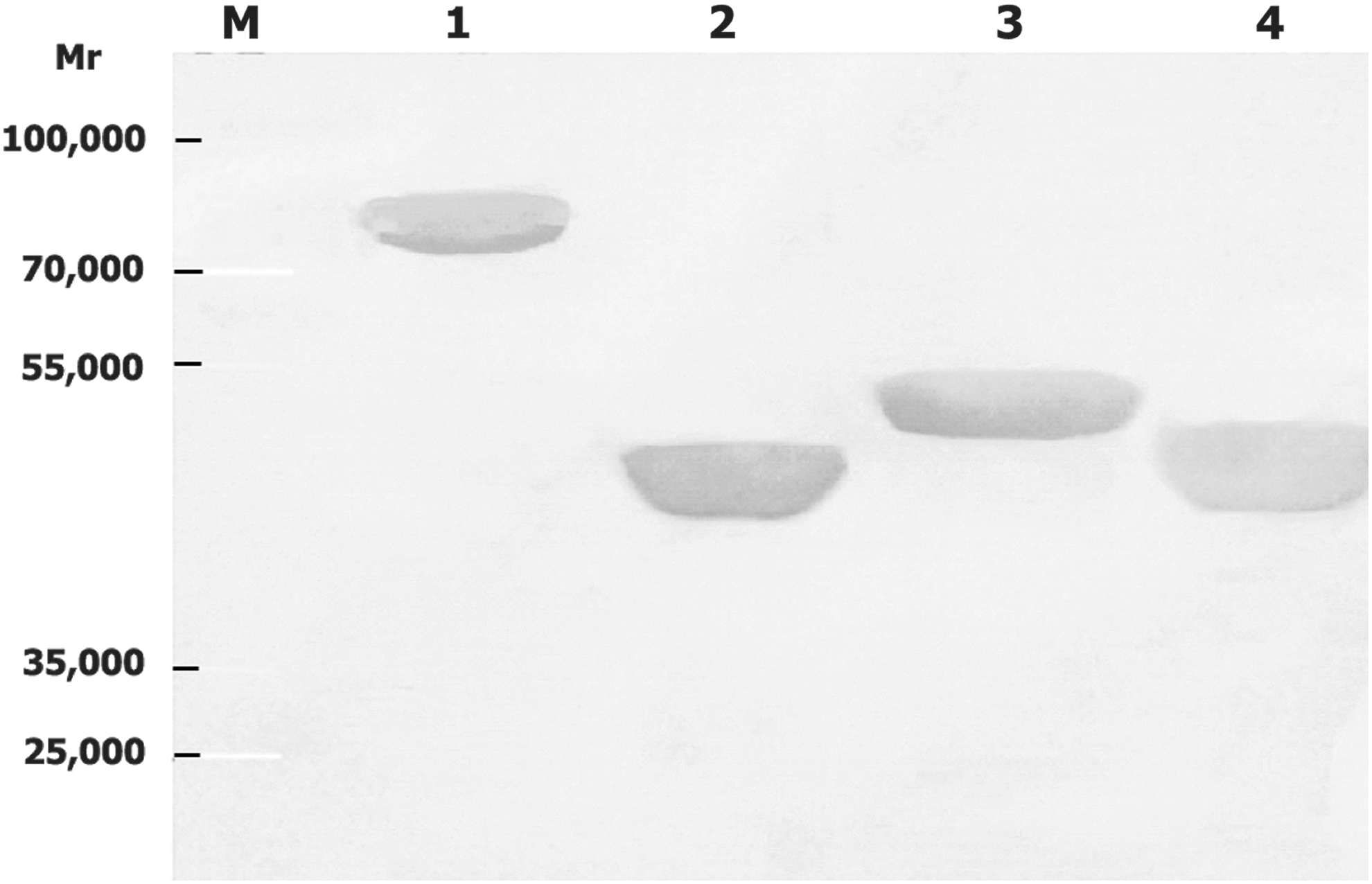
Western blot results showed that antibodies elicited in all the four guinea pigs in each group immunized with chimeric proteins carrying a single VP4 epitope reacted with the vector protein, VP6F, chimeric proteins carrying VP4 epitopes, recombinant VP4 protein, and the virus VP4 (about 88 kDa) and VP6 proteins (about 49 kDa) (Fig. 6, only one result is shown). No immunoreactivity of serum derived from pre-immune or negative control animals with VP6F, VP4 protein, or the chimeric proteins carrying VP4 epitopes were observed (data not shown).

Western blot of rotavirus proteins with guinea pig antibodies in guinea pig serum inoculated with chimeric protein(s) carrying VP4 epitope(s). Molecular weight standard (M), recombinant vector protein VP6F (1), chimeric proteins 6F/P[4]223 (2), 6F/P[8]296 (3), 6F/P[4]56 (4), recombinant VP4 protein (5), the virus VP4 and VP6 proteins of strain Wa (6), and SA11 (7). For each recombinant protein (lanes 1–5) about 4 μg was loaded. For virus Wa (lane 6) and SA11 (lane 7), about 3×106 TCID50 of was loaded.
Immuofluorescence analysis of RV antigen in SA11-infected MA104 cells
RV antigens synthesized in SA11-infected MA104 cells were detected by immunofluorescence assay using guinea pig antibodies against the chimeric proteins. The results showed that RV antigen in SA11 infected MA104 cells could be detected by the antibodies against SA11, VP6F, chimeric proteins (Fig. 7a–e). As control, no fluorescence was detected in SA11 infected cells when detected with negative control sera (Fig. 7f) or pre-inoculation sera (data not shown).
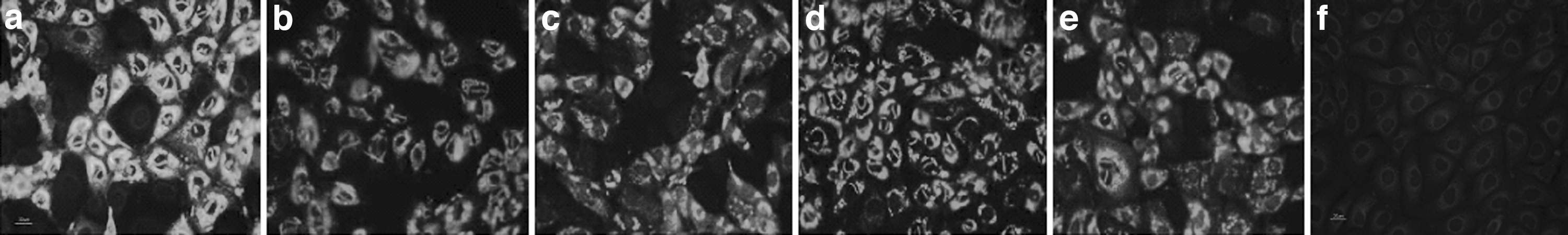
Immuofluorescence analysis of rotavirus antigens in SA11-infected MA104 cells with antibodies against SA11
Neutralizing antibodies activity
Neutralizing antibodies elicited in guinea pigs were determined. The results showed that antibodies in serum of all the guinea pigs inoculated with the recombinant chimeric proteins neutralized infectivity of Wa and SA11 in MA104 cells. Anti-rVP6F antibodies exhibited virus-neutralizing activity at lower titers (1:320). This level of neutralizing activity of anti-VP6F antibodies was the same as determined before for the antibodies against the recombinant native VP6 (date not shown). The neutralizing activity of antibodies against the recombinant chimeric proteins carrying VP4 epitopes was significantly higher than that against the VP6F protein. The neutralizing titers in antiserum from guinea pigs mock-inoculated with PBS as negative control or in pre-immune sera were lower than 1:10. The neutralizing titers in antisera from guinea pigs inoculated with the recombinant chimeric proteins that carrying VP4 epitopes were between 1:1280–1:5120 (Table 1).
Arithmetic mean NT titers; **NC, negative control. In this group, sera were derived from animals that were pre-immune or mock-immunized with PBS (see Materials and Methods).
Discussion
VP6 is highly conserved and is most abundant, constituting approximately 39% of the viral structure proteins by weight. The high degree of identity (>87%–99%) in the primary amino acid sequences of VP6 proteins from mammalian rotaviruses suggests VP6-based vaccines could potentially provide heterotypic protection. With specific properties that contain group antigenecity, capable of self-assembling into VLP (21), and potentially providing heterotypic protection, the VP6 was studied in attempts to develop VP6-based vaccines. Although some progress has been achieved, including using the VP6 as a vector for attachment/conjugation of foreign epitopes or others (13,34,43), and considered as a powerful carrier for displaying foreign epitopes (30), use of the native VP6 as an optimal vaccine or vector is still uncertain. First, the native VP6 lacks neutralizing antigenic contents belonging to the VP4 or VP7 as the major antigenic protein. Second, the native VP6 as a vector lacks proper insertion sites that can be conveniently used for insertion of foreign epitopes.
In the present study, using molecular cloning, genetic recombination, and expression techniques, a new foreign epitope presenting system based on RV VP6 as the vector was developed. Six sites that could be conveniently used for insertion of foreign epitopes were created n the outer surface of the vector protein,.
In construction of the system, the molecular stability of vector protein was carefully considered. In order to maintain the integrity of the vector protein conformation, all six insertion sites were created at the top of the loops of the protein that were exposed on the outer surface. The predicted molecular architecture showed that the backbone structure of VP6 was fully maintained in VP6F (Fig. 1B). Antigenicity of the native VP6 was another issue considered. The previous study showed that on the VP6 protein existed group-specific epitopes at the residual aa32-64, aa155-167, aa208-274, aa308-397 (17). All the group-specific epitopes were retained on the created vector protein.
With this system, three recombinant VP6-based VP4 epitope chimeric proteins were constructed. These chimeric proteins could react with anti-VP6 and anti-VP4 antibodies, and induced production of antibodies could recognize recombinant vector VP6F protein, recombinant VP4 protein, and the VP4 proteins of RV strain Wa or SA11. Antibodies in sera of guinea pigs immunized with recombinant VP6F or with the chimeric proteins carrying VP4 epitopes could neutralize infectious activity of Wa and SA11 in vitro.
Some VP4 epitopes had been described in previous studies. Among them, six peptides (aa1-10, aa35-44, aa55-66, and aa223-234, aa296-313, aa381-401) contained sequential antigenic determinants and were cross-reacting neutralization epitopes (18,19,38). These findings indicated that these sequential epitopes may also be important for the RV recombinant epitope chimeric vaccines. In the present study, three epitopes derived from the VP4 protein corresponding to the above mentioned epitopes were selected to construct recombinant epitope chimeric vaccines. These three epitopes were selected because they were sequential and comparably conserved. The results indicated that these epitopes had good immunoreactivity and enhanced imuunogenicity of the vector VP6F protein.
It was notable that antibodies produced in guinea pigs inoculated with the recombinant chimeric proteins had higher neutralizing titers against infectivity of SA11 than Wa. This might reflect the difference of the amino acid identity of the epitopes and the VP6 protein between SA11 and Wa (Table 2). For the epitope P[4]56 sequence, only one amino acid is different at the position aa64 between the P[4] (TB-Chen) and P[8] (Wa) and/or P[2] (SA11). Epitope P[4]223 sequences are identical in P[4] (TB-Chen), P[8] (Wa), and P[2] (SA11). Epitope P[8]296 sequences are comparably variable among P[4] and P[8], and P[2]. For the sequence of the VP6, percentage of amino acid identity between strains TB-Chen and SA11 (96.73%) is higher than that between strains TB-Chen and Wa (91.69%) (6).
Asterisks indicate identity with top line sequence. Amino acid substitutions are indicated.
It should be noted that only the epitopes derived from VP4 were constructed in the chimeric recombinant vaccines in this study; the chimeric recombinant vaccines carrying epitopes from VP7 should be further addressed. If it were confirmed that the chimeric recombinant vaccines using VP6F as a vector that carry epitopes from VP4 and VP7 elicit broadly protective immunity against diverse RVs in humans, it would represent potential second-generation vaccine candidates (the work about this will be described elsewhere). This is the first report of the RV VP6-based foreign epitope presenting system with six sites that can be conveniently used for insertion of six appropriate foreign epiotpes. It is expected that this VP6-based epitope presenting system and the recombinant VP6-based VP4 epitope chimeric proteins will be valuable for the development of novel RV vaccines and vaccine vectors.
Footnotes
Acknowledgments
The authors thank Chen Guo for technical assistance in construction of the VP6F molecular structure.
This work was supported in part by the Foundation of the Applied Basic Research Project of Yunnan Province, 2013FZ130, and the Foundation of the Applied Basic Research Project of Yunnan Province Youth Program, 2012FD039.
Author Disclosure Statement
The authors have no conflicts of interest.