
Abstract
CD8+ T-cells of asymptomatic HIV-1 carriers (AC) suppress human immunodeficiency virus type 1 (HIV-1) replication in a class I major histocompatibility complex (MHC-I)-restricted and -unrestricted manner. In order to investigate the mechanism of MHC-I-unrestricted CD8+ T-cell-mediated HIV-1 suppression, we previously established allo-antigen stimulated CD8+T-cells from HIV-1-uninfected donors. These allo-antigen stimulated CD8+ T-cells suppressed HIV-1 replication in acutely infected autologous CD4+ T-cells when directly co-cultured. To elucidate the mechanism of HIV-1 replication suppression, we analyzed DNA-binding activity and phosphorylation of transcriptional factors associated with HIV-1 replication by electrophoresis mobility shift assay and Western blotting. When CD4+ T-cells were cultured with allo-antigen stimulated CD8+ T-cells, the reduction of NF-κB and Ets-1 DNA-binding activity was observed. Nuclear localization of NF-κB p65 and Ets-1 was suppressed in CD4+ T-cells. Although NF-κB p65 and Ets-1 are known to be regulated by protein kinase A (PKA), no difference was observed in the expression and phosphorylation of the PKA catalytic subunit in CD4+ T-cells cultured with PHA-treated CD8+ T-cells or allo-antigen stimulated CD8+ T-cells. Cyclic AMP is also known to enter through gap junctions, but the suppression of HIV-1 replication mediated by allo-antigen stimulated CD8+ T-cells was not affected by the gap junction inhibitor. The nuclear transport of phosphorylated NF-κB p65 (Ser276) was inhibited only in CD4+ T-cells cultured with allo-antigen stimulated CD8+ T-cells. Our results indicate that allo-antigen stimulated CD8+ T-cells suppress the transcriptional activity of NF-κB p65 or Ets-1 in an antigen-nonspecific manner, and inhibit the nuclear transport of phosphorylated NF-κB p65 (Ser276).
Introduction
H
It is well known that the antiviral activities of AC-CD8+ T-cells involve both MHC-I-restricted and -unrestricted suppression (29). Previous studies have revealed that HIV-1-infected cells may be resistant to HIV-1-specific CTL-mediated cytotoxicity because of down-modulation of MHC-I by HIV-1 Nef (7). CTL activity against HIV-1-infected CD4+ T-cells may not be the only mechanism of CD8+ T-cell-mediated suppression. CD8+ T-cells produce many soluble factors that can suppress HIV-1 replication in vitro, including macrophage inflammatory protein 1-α (MIP-1α), MIP-1β, regulated on activation normal T-cell expressed and secreted (RANTES), stromal cell-derived factor 1, macrophage-derived chemokine (MDC), interleukin-16, interferons, and defensin (1,6,27,30,48,50). However, neutralizing antibodies against these cytokines or chemokines only partially inhibit the suppressive activity of CD8+ T-cells of AC (3,5,28). Levy et al. reported that CD8+ T-cell antiviral factor (CAF) can suppress HIV-1 replication at the transcriptional level without causing cell killing (22). However, the molecular aspects of CAF remain unclear. Furthermore, Liu et al. reported that HIV-1-irrelevant CD8+ CTLs established from HIV-1-uninfected donors also inhibit X4 and R5 HIV-1 replication in a cell-contact manner (21). These CTLs partially kill HIV-1-infected PBMC via the Fas ligand, but CTLs are able to suppress HIV-1 replication at a late stage in the presence of neutralizing antibodies against Fas ligand. The precise mechanisms of HIV-1 suppression by CD8+ CTLs are not fully understood. HIV-1 suppression may occur through regulation of transcriptional factors in CD4+ T-cells co-cultured with HIV-1-irrelevant CD8+ CTLs.
Previous papers have reported that protein kinase A (PKA) activity regulates both NF-κB transcriptional activity and Ca2+/calmodulin-dependent protein kinase II (CaMKII) activity that is able to control Ets-1 phosphorylation (19,24,38). Moreno-Fernandez et al. reported that cyclic AMP (cAMP) is transferred from regulatory T-cells to HIV-1 infected CD4+ T-cells through the connexin 43-gap junction (26). PKA activation by cAMP participates in HIV-1 suppression.
In the present study, we demonstrated that allo-antigen stimulated CD8+ T-cells are able to suppress both NF-κB and Ets-1 DNA binding activities in autologous CD4+ T-cells. The phosphorylated PKAc, Akt, and p38 MAPK were not affected in CD4+ T-cells cultured with allo-antigen stimulated CD8+ T-cells. Allo-antigen stimulated CD8+ T-cells treated with a gap junction inhibitor showed no change in HIV-1-suppressive activity and reduction of nuclear NF-κB p65 localization. Although NF-κB p65 (Ser276) in CD4+ T-cells was phosphorylated in CD4+ T-cells cultured with allo-antigen stimulated CD8+ T-cells, it was not transported from the cytoplasm to the nucleus. These findings indicate that allo-antigen stimulated CD8+ T-cells can suppress the transcriptional activity of both NF-κB and Ets-1, and inhibit the nuclear localization of phosphorylated NF-κB p65 (Ser276) in CD4+ T-cells.
Materials and Methods
Isolation of PBMC
This study was approved by the ethics committee in Kitasato University of Allied Health Sciences (2009-011). Informed consent was obtained from three healthy donors. PBMC were isolated by gradient centrifugation with Ficoll-Hypaque (Lymphoprep™; AXIS-SHIELD PoC, Oslo, Norway), and then stimulated overnight with 1% phytohemagglutinin-P (PHA-P’ Sigma-Aldrich, St. Louis, MO). The PBMC were then cultured in RPMI-1640 medium (Sigma-Aldrich) containing 10 U/mL recombinant human interleukin-2 (rhIL-2), 10% heat-inactivated fetal bovine serum (FBS; HyClone Laboratories, Inc., South Logan, UT), 100 U/mL penicillin, and 100 μg/mL streptomycin.
Induction of allo-antigen stimulated CD8+ T-cells
PBMC isolated from healthy donors were co-cultured with Raji cells (a human B-cell line) that had been treated with 0.05 mg/mL mitomycin C (MMC; Sigma-Aldrich) in RPMI-1640 with 10% heat-inactivated FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. Stimulation with MMC-treated Raji was performed every week. Four weeks later, CD8+ T-cells were separated from the cell culture by magnetic activating cell sorting (MACS; Miltenyi Biotec GmbH, Bergisch Gladbach, Germany). For flow-cytometric analysis, isolated CD8+ T-cells were stained using a combination of two fluorescence-conjugated monoclonal antibodies: CD3 (fluorescein isothiocyanate, FITC) and CD8 (phycoerythrin, PE; Becton Dickinson, Franklin Lakes, NJ). The stained cells were analyzed with an EPICS XL flow cytometer and EXPO 32™ software (Beckman Coulter, Miami, FL). Allo-antigen stimulated CD8+ T-cells established from healthy donors were designated NR8-, MR8-, and TR8-CD8+ T-cells.
CD4+ and CD8+ T-cell enrichment
CD4+ and CD8+ T-cell-enriched fractions were obtained by MACS separation from PHA-stimulated PBMCs originating from the same donor as that used for allo-antigen stimulated CD8+ T-cell induction. After separation, the enriched CD4+ T-cells were cultured with autologous enriched CD8+ T-cells and allo-antigen stimulated CD8+ T-cells at a ratio of 1:3. After 1 day and 4 days of culture, CD4+ T-cells were enriched for preparation of nuclear and cytoplasmic extracts.
Preparation of nuclear and cytoplasmic extracts from CD4+ T-cells
Nuclear and cytoplasmic extracts were prepared from CD4+ T-cells using a Nuclear/Cytosol Fractionation Kit (BioVision, Inc., Mountain View, CA). The protein concentration of nuclear extracts was measured by Bradford assay (Bio-Rad Laboratories, Inc., Hercules, CA). The protein concentration in the cytoplasmic extracts was measured using a 2D-Quant kit (GE Healthcare, Little Chalfont, United Kingdom).
Electrophoresis mobility shift assay
Electrophoresis mobility shift assay (EMSA) was performed using nuclear extracts from CD4+ T-cells; double-stranded DNA oligomers were labeled with [α-32P]-dNTP with a Klenow Fragment and Random Primer DNA Labeling Kit v2.0 (Takara Bio, Inc., Shiga, Japan). Table 1 shows the sequences of the double-stranded DNA oligomers used. For blocking of nonspecific DNA-protein binding, 1 μg of nuclear extract was incubated in reaction mixture including 20 mM HEPES-KOH (pH 7.9), 50 mM KCl, 5% glycerol, 1 mM EDTA (pH 8.0), 10 mM DTT, BSA (10 mg/mL), and poly dI-dC (1 mg/mL) for 15 min on ice. After incubation, [α-32P]-labeled DNA oligomers and/or cold-competitors were added to the reaction mixture for 20 min on ice. The reaction mixture was then loaded on a 5% polyacrylamide gel and electrophoresed in 0.5× TBE buffer containing 45 mM Tris, 45 mM boric acid, and 25 mM EDTA at 150 V at 4°C. The gel was dried, and DNA-protein complexes were detected by autoradiography.
Western blotting
Nuclear and cytoplasmic extracts from CD4+ T-cells were fractionated by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE). Western analysis was performed using the following-primary antibodies: anti-PKA-C rabbit monoclonal antibody, anti-PKA-C phospho-Thr197 rabbit monoclonal antibody, anti-NF-κB p65 mouse monoclonal antibody, anti-NF-κB p65 phospho-Ser276 rabbit polyclonal antibody, anti-Akt rabbit polyclonal antibody, anti-Akt phospho-Ser473 rabbit monoclonal antibody, anti-p38 MAPK rabbit polyclonal antibody, anti-p38 MAPK phospho-Thr180/Tyr182 rabbit monoclonal antibody, anti-histone H3 mouse monoclonal antibody (Cell Signaling Technology Japan, K.K., Tokyo, Japan), anti-Ets-1 mouse monoclonal antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), anti-GAPDH mouse monoclonal antibody, and anti-beta actin mouse monoclonal antibody (Abcam, Cambridge, United Kingdom). Each protein was detected using appropriate secondary antibodies: horseradish peroxidase (HRP) conjugated donkey anti-rabbit IgG polyclonal antibody (BioLegend, San Diego, CA), and HRP conjugated goat anti-mouse IgG (American Qualex, Inc., San Clemente, CA), and visualized on X-ray film (FujiFilm Corporation, Tokyo, Japan) after incubation with a chemiluminescent substrate (GE Healthcare Japan, Tokyo, Japan).
In vitro HIV-1 infection of CD4+ T-cells
HIV-1 used for in vitro infection was a filtered culture supernatant of MOLT4/LAI C-3 cells (11). For HIV-1 infection in vitro, CD4+ T-cells isolated from HIV-1 uninfected healthy donors were spinoculated with culture supernatants containing HIV-1 at a multiplicity of infection of 0.03 to 0.1 at room temperature for 1.5 h, extensively washed, and cultured in a medium containing recombinant human IL-2 at a concentration of 105 cells/200 μL in a 96-well round-bottom plate.
CD8+ T-cell treatment with a gap junction inhibitor
CD8+ T-cells were treated with connexin 43 mimetic peptide GAP 27, a gap junction inhibitor (100 μm; Sigma-Aldrich), for 2 h before co-culture with autologous CD4+ T-cells. Then, CD8+ T-cells were extensively washed, and co-cultured with HIV-1 infected CD4+ T-cells. After 4 days of incubation, the concentration of HIV-1 p24 antigen in the culture supernatants was measured using an enzyme-linked immunosorbent assay (ELISA; ELISA kits were kindly provided by Dr. Y. Tanaka, University of the Ryukyus. Japan).
Flow-cytometric analysis
PHA-treated CD8+ T-cells and established allo-antigen stimulated CD8+ T-cells were stained with the following fluorescent conjugated monoclonal antibodies: CD8 (FITC), VCAM-1 (PE), Fas-L (PE), BTLA (PE), 2B4 (PerCP/Cy5.5), PD-1 (PerCP/Cy5.5), Tim-3 (PE/Cy7), CTLA-4 (APC), and ICAM-1 (APC) (BioLegend). The stained cells were analyzed by a MACSQuant flow cytometer (Miltenyi Biotec).
Statistics
Statistical significance was analyzed using the Mann–Whitney U-test. Values were considered statistically significant when p<0.05.
Results
1. Suppression of HIV-1 replication and DNA-binding activity of transcriptional factors associated with HIV-1 replication by allo-antigen stimulated CD8+ T-cells
To study the mechanism of HIV-1 suppression by CD8+ T-cells, we investigated whether allo-antigen stimulated CD8+ T-cells established from healthy donors were able to suppress HIV-1 replication. Allo-antigen stimulated CD8+ T-cells efficiently suppressed HIV-1 replication from HIV-1 infected CD4+ T-cells, whereas no such suppressive activity was demonstrated by PHA-treated CD8+ T-cells (PHA8; Fig. 1A). To evaluate the influence of CD4+ T-cell death in the HIV-1-suppressive activity of allo-antigen stimulated CD8+ T-cells, we also examined whether uninfected CD4+ T-cells were killed by PHA8 and allo-antigen stimulated CD8+ T-cells. Neither cell death nor cleaved caspase-3 was evident in uninfected CD4+ T-cells cultured with both types of stimulated CD8+ T-cells (Supplementary Fig. S1A and B; Supplementary Data are available online at
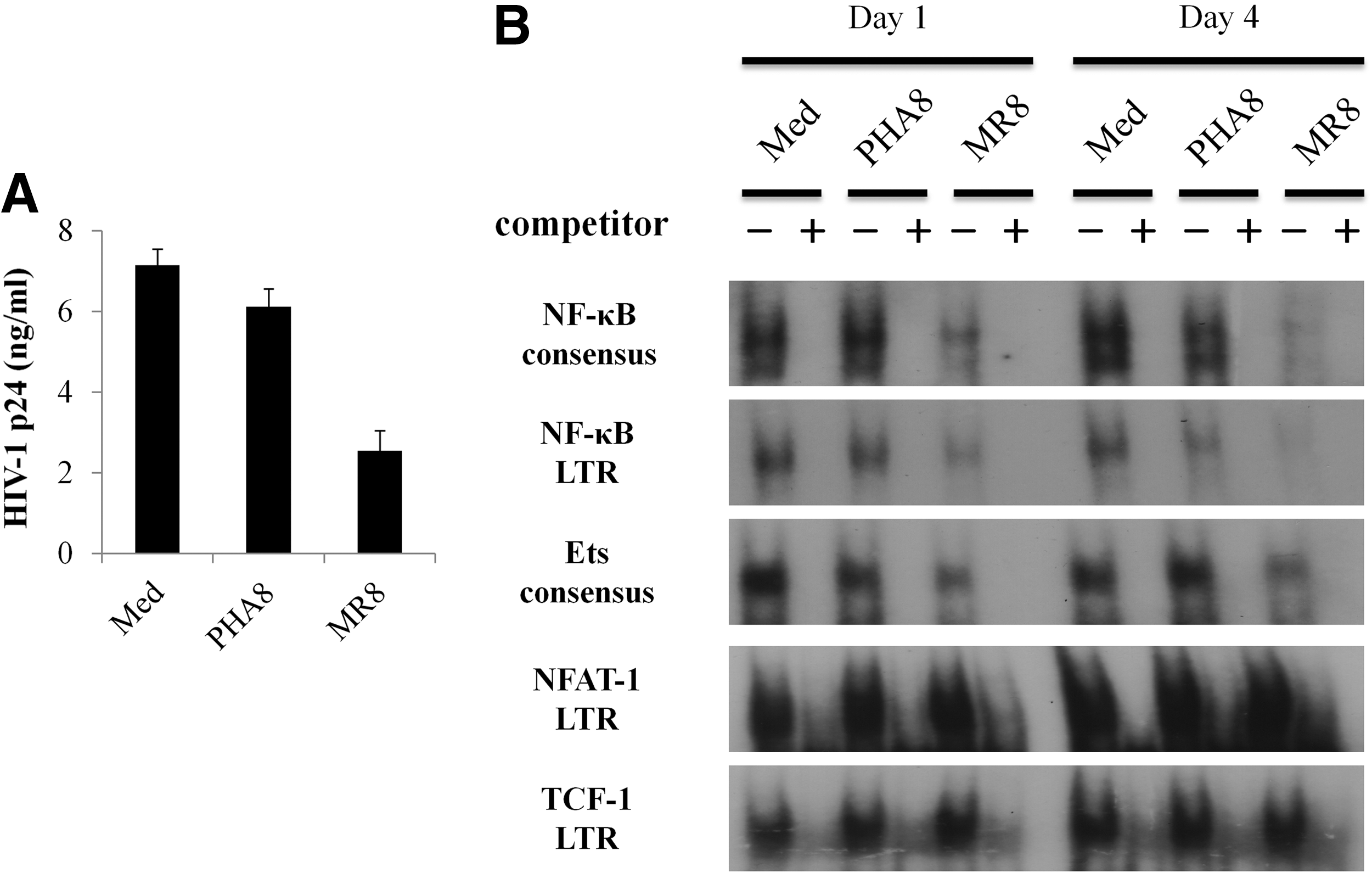
Suppression of human immunodeficiency virus type 1 (HIV-1) replication and transcriptional activity by allo-antigen stimulated CD8+ T-cells.
2. Nuclear localization of NF-κB p65 and Ets-1 in CD4+ T-cells
To investigate the cellular localization of NF-κB p65 and Ets-1, we extracted the nuclear and cytoplasmic fraction from uninfected CD4+ T-cells cultured with either PHA8 or allo-antigen stimulated CD8+ T-cells, and carried out Western blotting analysis. After 1 day and 4 days of incubation, nuclear localization of both NF-κB p65 and Ets-1 was significantly reduced in uninfected CD4+ T-cells cultured with allo-antigen stimulated CD8+ T-cells (Fig. 2A–C). In contrast, there was no reduction of nuclear localization of either NF-κB or Ets-1 in uninfected CD4+ T-cells cultured with PHA8 (Fig. 2A–C). There was no evident reduction of nuclear NF-κB and Ets-1 localization in uninfected CD4+ T-cells cultured with culture supernatants of PHA8 and allo-antigen stimulated CD8+ T-cells (Supplementary Fig. 1D). We also examined the nuclear localization of NF-κB p65 in uninfected CD4+ T-cells at early stages of culture with PHA8 and allo-antigen stimulated CD8+ T-cells. After 2 h of culture, there was no evident reduction of NF-κB p65 nuclear localization in uninfected CD4+ T-cells in the presence of either type of stimulated CD8+ T-cells (Supplementary Fig. S2A and B). These results indicated that allo-antigen stimulated CD8+ T-cells inhibited the nuclear translocation of both NF-κB p65 and Ets-1 in autologous CD4+ T-cells.
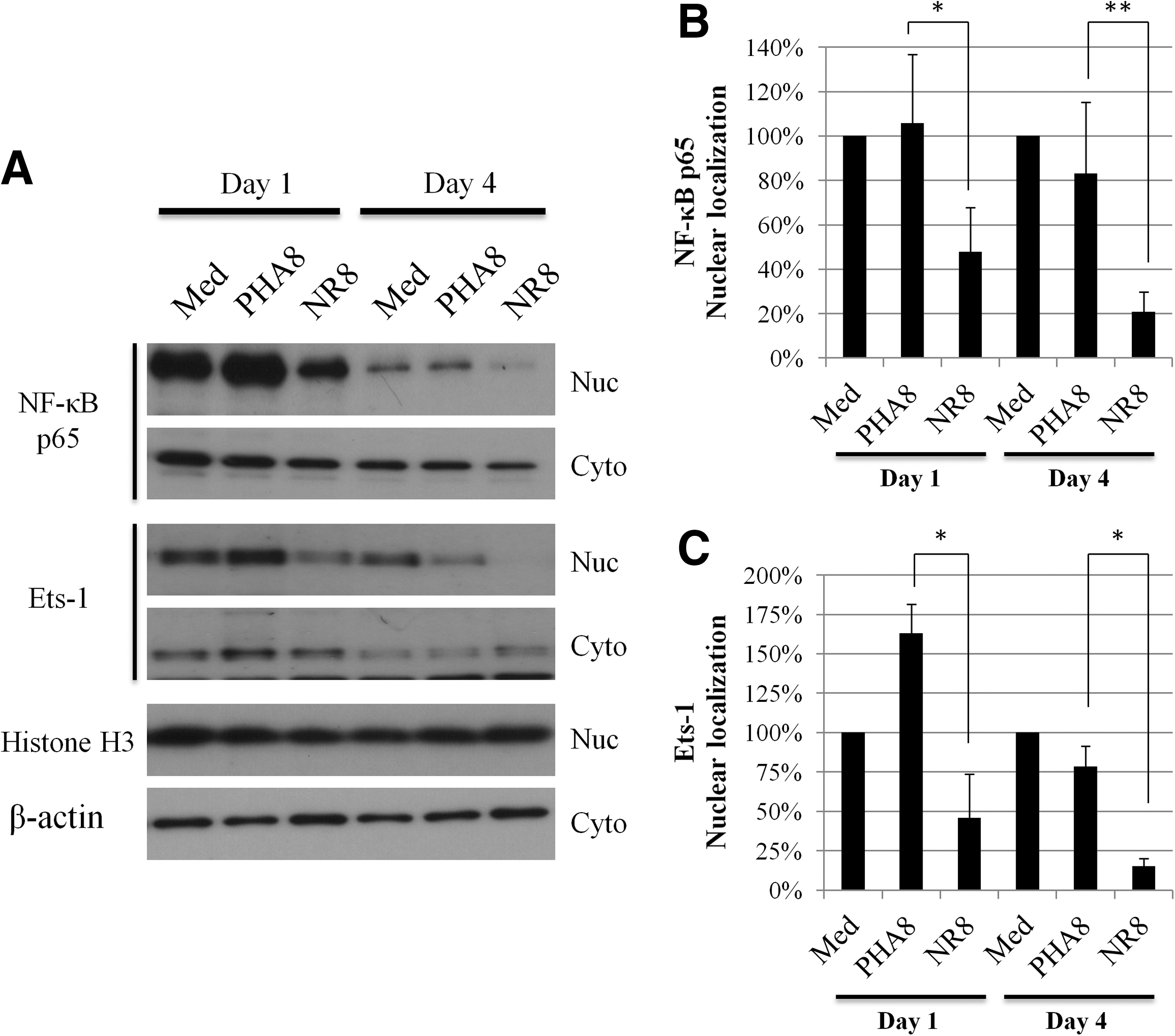
Localization of NF-κB or Ets-1 in uninfected CD4+ T-cells.
3. Participation of PKA, Akt, and p38 MAPK in the reduction of NF-κB and Ets-1 DNA-binding activities
Protein kinase A (PKA) activity regulates nuclear localization and NF-κB transcriptional activity. Ets-1 is also phosphorylated by Ca2+/calmodulin-dependent kinase II (CaMKII) through PKA. To investigate the participation of PKA in the HIV-1-suppressive activity by allo-antigen stimulated CD8+ T-cells, we carried out Western blotting analysis of the PKA catalytic subunit (PKAc) and phosphorylated PKAc (Thr197) in uninfected CD4+ T-cells cultured with PHA8 and allo-antigen stimulated CD8+ T-cells. Neither PHA8 nor allo-antigen stimulated CD8+ T-cells affected the amount of PKAc in uninfected CD4+ T-cells (Fig. 3A and B). We then assessed the phosphorylated PKAc (Thr197) in uninfected CD4+ T-cells, as PKA activity is regulated by PKAc phosphorylation (Thr197). However, phosphorylated PKAc (Thr197) was not affected in uninfected CD4+ T-cells cultured with PHA8 and allo-antigen stimulated CD8+ T-cells (Fig. 3A and C). We also examined the phosphorylated PKAc (Thr197) in uninfected CD4+ T-cells at early stages of culture with PHA8 and allo-antigen stimulated CD8+ T-cells. There was no evident reduction of phosphorylated PKAc (Thr197) in uninfected CD4+ T-cells after 2 h of culture with allo-antigen stimulated CD8+ T-cells (Supplementary Fig. S2C and D). Akt, also known as protein kinase B (PKB), and p38 mitogen-activated protein kinase (MAPK) are also known to regulate NF-κB signaling via activation of the IKK complex and induction of IκB degradation. To study the participation of Akt and p38 MAPK in HIV-1-suppressive activity, we further examined the phosphorylation of both Akt and p38 MAPK in uninfected CD4+ T-cells cultured with PHA8 and allo-antigen stimulated CD8+ T-cells. The amounts of phosphorylated Akt and p38 MAPK were not significantly affected in uninfected CD4+ T-cells under either of these culture conditions (Fig. 3D–F). Similarly, the amounts of unphosphorylated Akt and p38 MAPK were unaffected (data not shown). These results suggested that the reduction of NF-κB p65 and Ets-1 activities in uninfected CD4+ T-cells cultured with allo-antigen stimulated CD8+ T-cells might not be due to PKAc, Akt, and p38 MAPK activation.
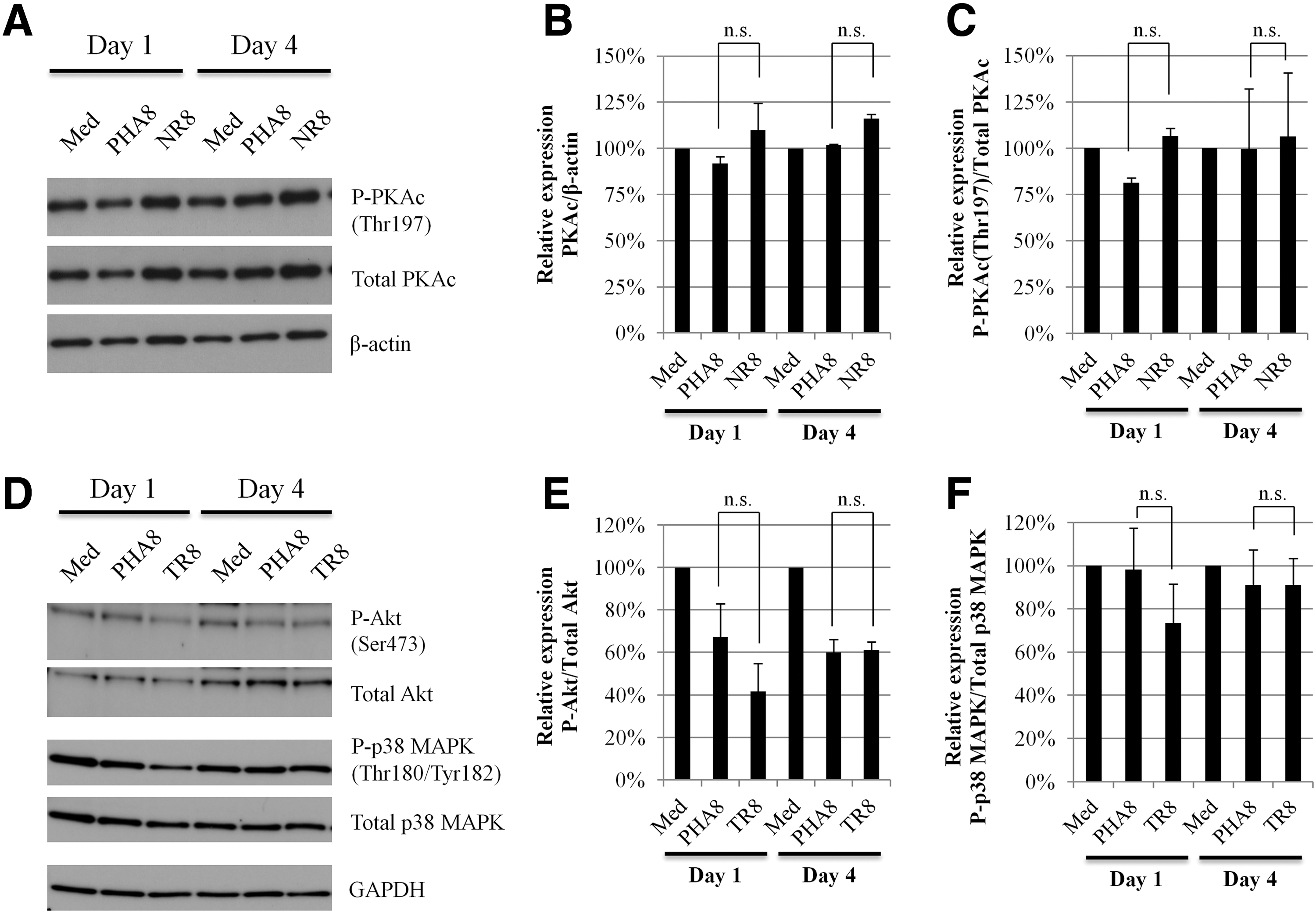
Analysis of PKAc, Akt, and p38 MAPK activity in the cytoplasm of uninfected CD4+ T-cells cultured with CD8+ T-cells.
4. Effects of the cAMP influx from allo-antigen stimulated CD8+ T-cells to CD4+ T-cells on HIV-1 replication and DNA-binding activity
Cyclic AMP (cAMP) can flow from cell to cell gap junctions, and activate PKA. To investigate the HIV-1 suppressive effect of cAMP influx, we examined whether HIV-1 replication was inhibited by transfer of cAMP through gap junctions from allo-antigen stimulated CD8+ T-cells to autologous HIV-1 infected CD4+ T-cells. To inhibit cAMP influx, PHA8 and allo-antigen stimulated CD8+ T-cells were treated with a connexin 43 gap junction inhibitor (GAP 27), as described in Materials and Methods. Although allo-antigen stimulated CD8+ T-cells suppressed HIV-1 replication in autologous HIV-1 infected CD4+ T-cells, GAP 27 treatment of allo-antigen stimulated CD8+ T-cells did not affect abrogation of the HIV-1 suppressive activity with allo-antigen stimulated CD8+ T-cells (Fig. 4A). At the same time, we also examined the nuclear localization and DNA-binding activity of NF-κB in uninfected CD4+ T-cells cultured with GAP 27-treated PHA8 and allo-antigen stimulated CD8+ T-cells. We also observed the reduction of NF-κB nuclear localization and DNA-binding activity in uninfected CD4+ T-cells cultured with GAP 27-treated allo-antigen stimulated CD8+ T-cells (Fig. 4B and C). These results suggested that HIV-1 suppression by allo-antigen stimulated CD8+ T-cells was not related to molecules involved in the passage of cAMP through connexin 43 gap junctions.
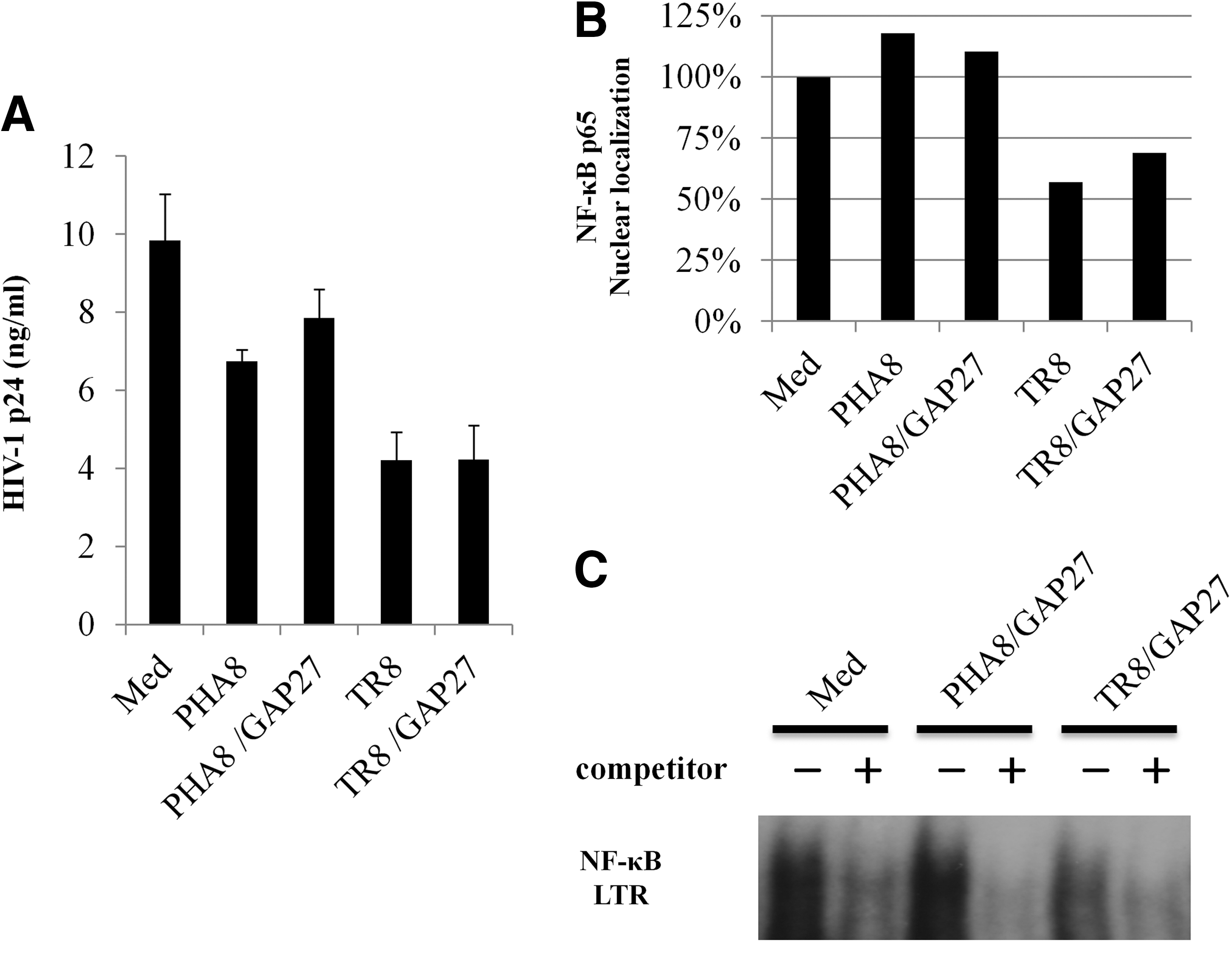
Participation of gap junctions in HIV-1 suppression by allo-antigen stimulated CD8+ T-cells.
5. Cellular localization of phosphorylated NF-κB p65 (Ser276) in CD4+ T-cells
As described above, NF-κB p65 in uninfected CD4+ T-cells cultured with allo-antigen stimulated CD8+ T-cells localized more in the cytoplasm than in the nucleus (Fig. 2A). NF-κB nuclear translocation occurs after IκBα, which masks nuclear localization signal of NF-κB p65, is degraded by proteasomes. Phosphorylation of NF-κB p65 by a number of kinases enhances NF-κB transcriptional activity and nuclear localization. To clarify the state of nuclear phosphorylated NF-κB p65, we investigated whether NF-κB p65 in the cytoplasm and nucleus of uninfected CD4+ T-cells cultured with PHA8 and allo-antigen stimulated CD8+ T-cells was phosphorylated at serine residues 276 and 536. Phosphorylated NF-κB p65 (Ser276) in the cytoplasm was observed only in uninfected CD4+ T-cells cultured with allo-antigen stimulated CD8+ T-cells, whereas it was not detected in uninfected CD4+ T-cells cultured with PHA8 (Fig. 5A). We observed less phosphorylated NF-κB p65 (Ser276) in the nuclei of CD4+ T-cells cultured with allo-antigen stimulated CD8+ T-cells than in those cultured with PHA8 for 1 day. After 4 days of culture, phosphorylated NF-κB p65 (Ser276) was not clearly detectable in the nuclei of uninfected CD4+ T-cells cultured with allo-antigen stimulated CD8+ T-cells (Fig. 5A and B), whereas it was transported from the cytoplasm to the nucleus in uninfected CD4+ T-cells cultured with PHA8 (Fig. 5A and B). Phosphorylated NF-κB p65 (Ser276) was only retained in the cytoplasm of uninfected CD4+ T-cells cultured with allo-antigen stimulated CD8+ T-cells (Fig. 5A and B), but the total amounts of phosphorylated NF-κB p65 (Ser276) and NF-κB p65 in uninfected CD4+ T-cells did not differ, irrespective of culture with PHA8 and allo-antigen stimulated CD8+ T-cells (Fig. 5A and C). In contrast, there was no difference in the amount of cytoplasmic phosphorylated NF-κB p65 (Ser536) in uninfected CD4+T-cells, irrespective of culture with PHA8 and allo-antigen stimulated CD8+ T-cells (Supplementary Fig. S3). These results indicated that although NF-κB p65 was phosphorylated in CD4+ T-cells cultured with PHA8 and allo-antigen stimulated CD8+ T-cells, nuclear transport of a proportion of phosphorylated NF-κB p65 (Ser276) was inhibited by an unknown HIV-1 suppressive mechanism potentially related to HIV-1 pathogenesis/replication by allo-antigen stimulated CD8+ T-cells.
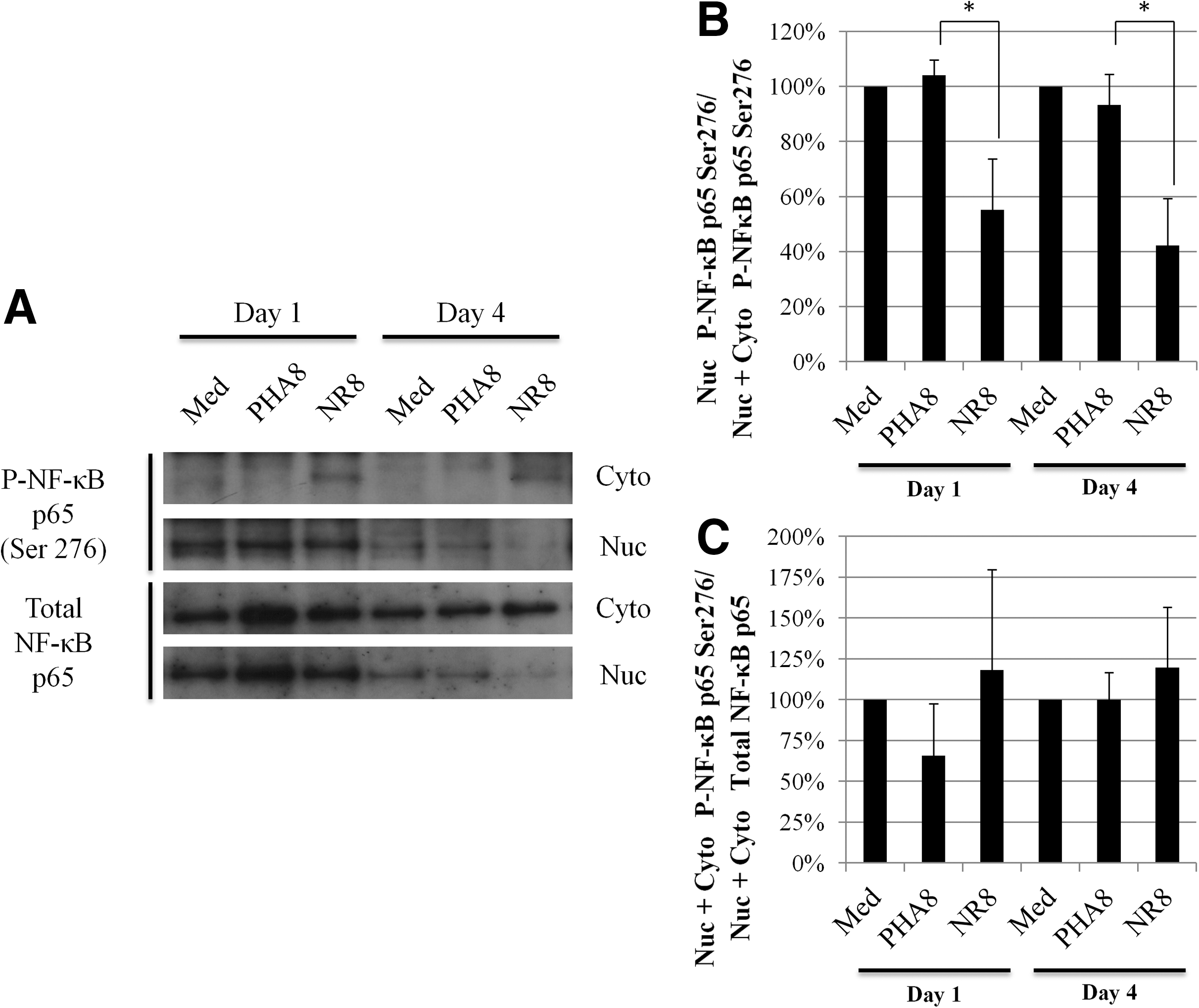
Analysis of phosphorylated NF-κB p65 Ser276 in uninfected CD4+ T-cells.
6. Flow cytometric analysis of allo-antigen stimulated CD8+ T-cells
To study the differences in HIV-1-suppressive activity between PHA8 and allo-antigen stimulated CD8+ T-cells, we compared the surface expression of costimulatory molecules, adhesion molecules, and negative regulatory molecules on CD8+ T-cells. Neither cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) nor Fas-ligand (Fas-L) was expressed on either PHA8 or allo-antigen stimulated CD8+ T-cells, whereas the expression of B and T lymphocyte attenuator (BTLA) tended to be slightly higher on the former than on the latter. Expression of vascular cell adhesion molecule 1 (VCAM-1) was slightly higher on allo-antigen stimulated CD8+ T-cells than PHA8, whereas that of intercellular adhesion molecule 1 (ICAM-1) was significantly higher on the former. Allo-antigen stimulated CD8+ T-cells showed higher expression of 2B4, programmed death-1 (PD-1), and T-cell immunoglobulin domain and mucin domain-3 (Tim-3; Fig. 6 and Table 2). These results indicated that CD8+ T-cells stimulated with allo-antigen might be more exhausted than those stimulated with PHA, but that higher expression of ICAM-1 on allo-antigen stimulated CD8+ T-cells might strengthen cell-to-cell contact between autologous CD4+ T-cells and allo-antigen stimulated CD8+ T-cells.
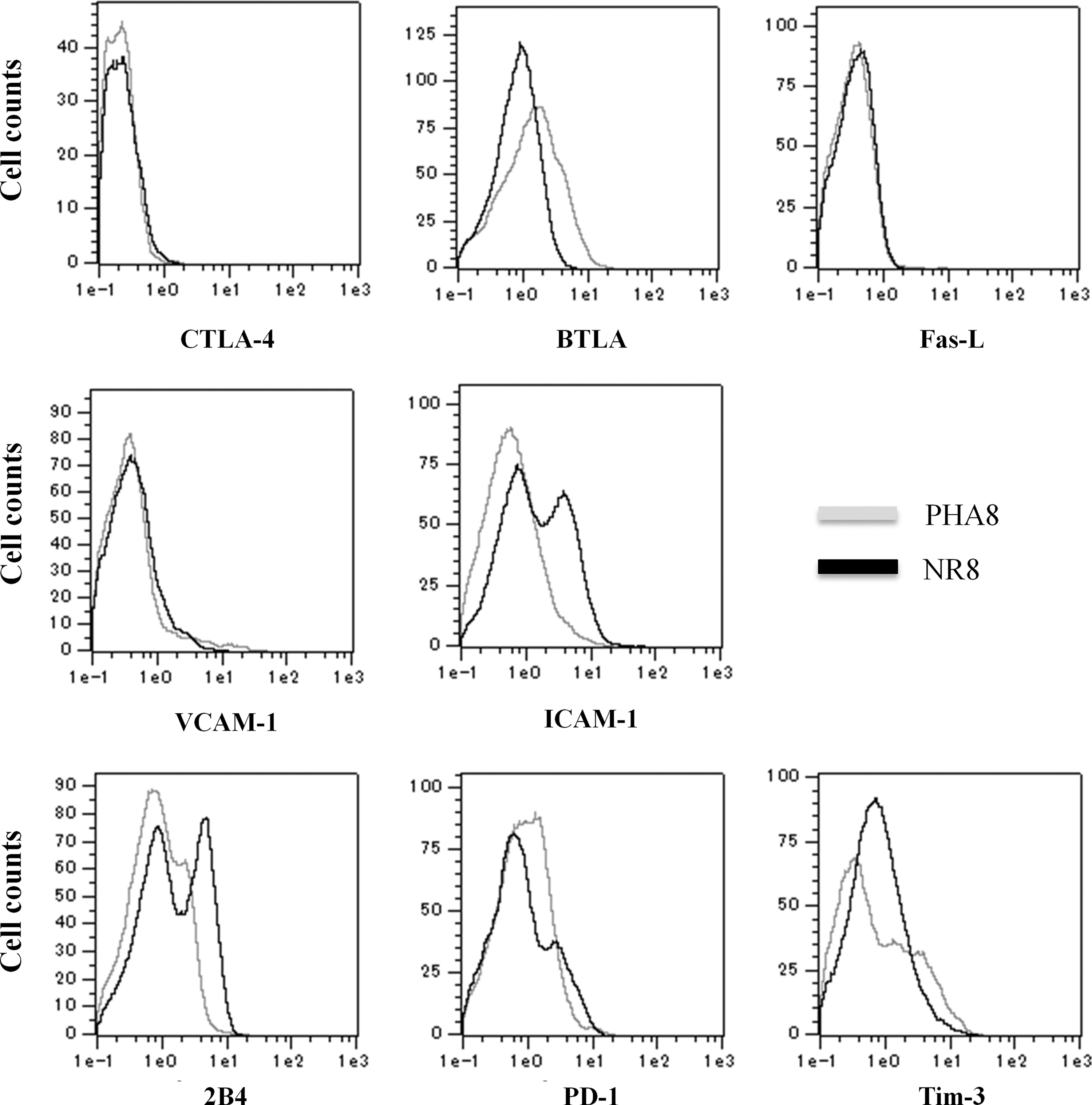
Characterization of allo-antigen stimulated CD8+ T-cells. Allo-antigen stimulated CD8+ T-cells (black line) and PHA-treated CD8+ T-cells (gray line) derived from three healthy donors were characterized by flow-cytometric analysis using immunofluorescent antibodies staining for CD8, CTLA-4, BTLA, Fas-L, VCAM-1, ICAM-1, 2B4, PD-1, and Tim-3. The data for one of five independent experiments are shown.
The results indicate the average±SD of five independent experiments. ** p<0.01.
PHA8, PHA-treated CD8+ T-cells; Allo8, allo-antigen stimulated CD8+ T-cells.
Discussion
In the present study, we showed that allo-antigen stimulated CD8+ T-cells established from uninfected healthy donors induced a reduction of DNA binding activity of NF-κB and Ets-1 in uninfected CD4+ T-cells in vitro (Fig. 1). Nuclear localization of NF-κB p65 and Ets-1 was reduced in uninfected CD4+ T-cells cultured with allo-antigen stimulated CD8+ T-cells (Fig. 2). We confirmed that the HIV-1-suppressive activity of allo-antigen stimulated CD8+ T-cells was caused by reduction of NF-κB p65 and Ets-1 in the nucleus. Levy et al. reported that CD8+ CAF produced by CD8+ T-cells of asymptomatic carriers is able to suppress HIV-1 replication at the transcriptional level without causing cell killing (22). Le Borgne et al. previously reported that Epstein-Barr virus-specific CTLs from an HIV-seronegative donor suppressed HIV-1 replication (17). Moreover, previous reports have shown that HIV-1-irrelevant allo-antigen stimulated CD8+ T-cells established from healthy donors are able to suppress HIV-1 viral replication at the late stage (16,21). We demonstrated that cell viability and the reduction of nuclear NF-κB p65 and Ets-1 localization in CD4+ T-cells were not influenced by culture with culture supernatants of PHA-treated and allo-antigen stimulated CD8+ T-cells (Supplementary Fig. S1). Our results are in accord with these reports, and we were able to identify the transcriptional factors, NF-κB and Ets-1, as key players in HIV-1 suppression mediated by allo-antigen stimulated CD8+ T-cells. Activated CD8+ T-cells established by allo-antigen stimulation might have functions for NF-κB- and Ets-1-mediated reduction of both DNA-binding activity and nuclear localization.
Akt and p38 MAPK regulate NF-κB signaling via the activation of IKK complex and the induction of IκB degradation (23,36,40). Moreover, PKA activity regulates NF-κB transcriptional activity through NF-κB p65 (Ser276) phosphorylation (38). NF-κB p65 (Ser276) phosphorylation upregulates NF-κB transcriptional activity through various effects including recruitment of cAMP response element binding protein (CREB) binding protein (CBP)/p300, which is a transcriptional co-activator (9,51,52). A previous study has shown that cAMP negatively regulates HIV-1 enhancer activity and virus production. CREB protein activated by cAMP-dependent PKA activity competes with phosphorylated NF-κB and Ets-1 proteins for limiting amounts of CBP/p300 (2). Therefore, we focused on PKA activity because PKA is able to control both NF-κB p65 and Ets-1 transcriptional activity. In the present study, however, we found no differences of phosphorylated PKAc (Thr197), Akt (Ser473), and p38 MAPK (Thr180/Tyr182) in uninfected CD4+ T-cells cultured with PHA8 and allo-antigen stimulated CD8+ T-cells (Fig. 3). Previously, Moreno-Fernandez et al. reported that regulatory T-cells also control HIV-1 replication in autologous CD4+ T-cells by cAMP-dependent PKA activation through gap junctions (26). We examined whether allo-antigen stimulated CD8+ T-cells have the HIV-1 suppressive mechanism that is similar to regulatory T-cells. However, treatment of allo-antigen stimulated CD8+ T-cells with GAP 27 did not affect HIV-1 replication in autologous CD4+ T-cells (Fig. 4). We considered that the reduction of NF-κB p65 and Ets-1 nuclear localization might not be regulated by the difference of PKAc in uninfected CD4+ T-cells cultured with PHA8 and allo-antigen stimulated CD8+ T-cells.
Interestingly, the phosphorylated NF-κB p65 (Ser276) was only retained in the cytoplasm of uninfected CD4+ T-cells cultured with allo-antigen stimulated CD8+ T-cells (Fig. 5). The A kinase interacting protein 1 (AKIP1) binds to NF-κB p65 and regulates its transcriptional activity through phosphorylation at serine residue 276 by PKAc (14). Nuclear accumulation of NF-κB p65 can be delayed by replacing serine residue 276 with a phosphomimetic. Although the contribution of AKIP1 is unknown in our study, AKIP activity might play a role in the HIV-1-suppressive mechanism in CD4+ T-cells cultured with allo-antigen stimulated CD8+ T-cells.
It has been reported that NF-κB signaling is negatively regulated by inhibition of DNA-binding function, IKK activity, and IκB degradation. Protein inhibitor of activated STAT1 (PIAS1) directly interacts with NF-κB p65 and prevents it from binding to DNA (20). Another member of the PIAS family, PIASy, also negatively regulates both STAT1 and NF-κB signaling by cooperating with PIAS1 (37). Fas-associated Factor-1 (FAF-1) inhibits the nuclear transport of NF-κB p65. FAF-1 disrupts IKK complex formation through physical interaction with the leucine-zipper domain of IKKβ, and attenuates IKKγ recruitment to IKKβ. As a result, FAF-1 suppresses NF-κB activation by inhibiting the nuclear localization of NF-κB p65 (31,32). Another study has shown that caveolin 1 (Cav-1), the scaffold protein of a specific membrane lipid raft known as a caveola, is also expressed in T lineage cells, dendritic cells, and monocytes/macrophages (39,46). Cav-1 suppresses HIV-1 replication and decreases the translocation of NF-κB p65 to the nucleus in Cav-1-overexpressing cell lines and macrophages (46). A20 is also one of the major negative regulators of NF-κB signaling, mediating the removal of K63-polyubiquitin chains from receptor interacting protein 1 (RIP1), and catalyzing RIP1 K48-polyubiquitination, thus allowing RIP1 to be degraded by proteasomes (47). Moreover, TAXBP1, which was first identified as a protein that interacts with HTLV Tax protein, facilitates the interaction of A20 with RIP1 (35). A20 binding inhibitors of NF-κB (ABIN) interacts with A20 and also negatively regulates NF-κB signaling (41,42). It has also been reported another member the ABIN family, ABIN-2, markedly reduces hepatectomy-induced NF-κB p65 nuclear localization in hepatocytes of ABIN-2 transgenic mice, relative that that in wild-type mice (18). Various molecules expressed on allo-antigen stimulated CD8+ T-cells are likely to influence signal transduction through PIAS1, FAF-1, Cav-1, A20, ABINs, TAXBP1, or AKIP1 in CD4+ T-cells expressing receptor molecule(s). The nature of these molecules remains to be clarified.
In investigating whether some molecules on CD8+ T-cells are associated with suppression of HIV-1 replication, we found marked differences of 2B4, Tim-3, and ICAM-1 expression on allo-antigen stimulated CD8+ T-cells (Fig. 6 and Table 2). Yamamoto et al. reported that the negative regulatory molecules PD-1, CD160, and 2B4 are more highly expressed on HIV-specific CD8+ T-cells before antiretroviral therapy (49). These molecules are highly expressed on the surface of exhausted CD8+ T-cells. Our established allo-antigen stimulated CD8+ T-cells showed high expression of 2B4 and Tim-3, and slight expression of PD-1. This might suggest that CD8+ T-cells stimulated with allo-antigen might be more exhausted than those stimulated with PHA. Diaz et al. have reported that VCAM-1 expression on CD8+ T-cells correlates with enhanced anti-HIV-1 suppression activity (8). They showed that VCAM-1+CD8+ T-cells produce an anti-HIV soluble factor, such as CAF, but not VCAM-1, as anti-VCAM-1 antibody had no effect on the noncytotoxic anti-HIV response of CD8+ cells. Our established allo-antigen stimulated CD8+ T-cells tended to show slightly higher cell surface expression of VCAM-1 than CD8+ T-cells stimulated with PHA. However, we also demonstrated variation of VCAM-1 expression on allo-antigen stimulated CD8+ T-cells. Although VCAM-1 might play a role in the HIV-1-suppressive activity of CD8+ T-cells, it may not be the main player in the HIV-1-suppressive activity observed in the present system. Although our allo-antigen stimulated CD8+ T-cells showed high expression of ICAM-1, it is not clear whether ICAM-1 possesses direct HIV-1-suppressive activity. ICAM-1 may strengthen the response of CD4+ T-cells to contact with allo-antigen stimulated CD8+ T-cells.
In this study, we have identified the key transcriptional factors, NF-κB p65 and Ets-1, related to HIV-1 suppression at the transcriptional level by allo-antigen stimulated CD8+ T-cells. Furthermore, this suppressive mechanism by allo-antigen stimulated CD8+ T-cells inhibits the nuclear transport of NF-κB p65, including phosphorylated NF-κB p65. In a future study, we intend to identify the factor(s) or mechanism(s) inhibiting the nuclear transport of phosphorylated NF-κB p65 (Ser276). If this HIV-1-suppressive mechanism(s) by allo-antigen stimulated CD8+ T-cells can be clarified, it will contribute to the development of therapeutic agents for HIV-1 and a new strategy for therapeutic vaccines.
Footnotes
Acknowledgments
We thank Dr. Mari Kannagi (Tokyo Medical and Dental University, Japan) for kindly providing MOLT4/LAI C-3 cells, and Dr. Yuetsu Tanaka (University of the Ryukyus, Japan) for kindly providing HIV-1 p24 ELISA kit. This study was supported by the Graduate School of Medical Sciences, Kitasato University (Integrative Research Program, 2008–2009, 2013), by Kitasato University Research Grant for Young Researchers (Grant-in Aid 2008), and the Kanagawa Nanbyou Study Foundation (Grant-in Aid 2007).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.