
Abstract
Enterovirus 71 (EV71) infection can cause severe disease and lead to death in children. Recurring outbreaks of EV71 have been reported in several countries. Interferons (IFNs) have been used for decades to treat several types of viral infection, but have a limited ability to inhibit EV71 replication. Herein, we intend to investigate the mechanisms by which EV71 inhibits the cellular type I IFN response. In this study, MRC-5 (human embryonic lung fibroblast) or RD (human rhabdomyosarcoma) cells were infected with EV71, and then treated with or without IFN-α2b. Cells were harvested and analyzed by flow cytometry to determine the level of IFNAR1. Cell lysis were prepared to detect the levels of STAT1, STAT2, phosphorylated STAT1, phosphorylated STAT2, IFNAR1, JAK1, and TYK2 by Western blotting. The phosphorylation of STAT1 and STAT2 induced by IFN were inhibited without significant downregulation of IFNAR1 in EV71-infected cells. The EV71-induced suppression of STAT1 and STAT2 phosphorylation was not rescued by the protein tyrosine phosphatases inhibitor, and was independent of suppressor of cytokine signaling protein 1/3 levels. The phosphorylation of JAK1 and TYK2 were inhibited accompanied by EV71-induced downregulation of JAK1, which occurred at a post-transcriptional level and was proteasome independent. JAK1 expression did not decrease, and IFN-α-stimulated STAT1 and STAT2 phosphorylation were not blocked in HEK293T cells overexpressing the EV71 viral protein 2A or 3C. This study demonstrates that EV71 inhibits the cellular type I IFN antiviral pathway by downregulating JAK1, while the expression of IFNAR1 does not significantly alter in EV71-infected cells. Additionally, the EV71 viral proteins 2A and 3C do not act as antagonists of cellular type I IFN signaling.
Background
E
Type I interferon (IFN) is the first line of cellular defense against viral infection, and plays a vital role in host innate immunity through the induction of a variety of antiviral effects. Host cells are stimulated to induce an antiviral response by type I INF through the Janus activated kinase (JAK)-signal transducer and activation of transcription (STAT) signaling pathway (9,28,30,31). In brief, type I IFNs bind to type I IFN receptor on host cells that is composed of two subunits—IFNAR1 and IFNAR2—which are associated with the JAKs, tyrosine kinase 2 (TYK2), and JAK1 respectively. The conformational changes induced in IFNAR1 and IFNAR2 by type I IFNs lead to the activation of TYK2 and JAK1, and in turn, their activation results in the phosphorylation of STAT1 and STAT2. Phosphorylated STAT1 and STAT2 dimerize and bind to IFN-regulatory factor 9 (IRF9) to form IFN-stimulated gene factor-3 (ISGF3) complexes. These complexes translocate to the nucleus and bind to IFN-stimulated response elements (ISREs) to initiate the transcription of ISGs. These antiviral genes play a very important role in establishing the antiviral response in host cells (14).
Clinical and laboratory studies have shown that there is a limited response to type I IFNs in EV71-infected patients, which has prompted intense research into the mechanisms by which EV71 circumvents the type I IFN response. Lu et al. reported that EV71 blocked type I IFN signaling by reducing the cellular expression of IFNAR1 (18). In this study, we aimed to investigate further the mechanisms by which EV71 inhibits the cellular type I IFN response. In contrast to Lu et al. (18), our work suggests that EV71 inhibits type I IFN signaling by degrading JAK1, rather than altering the expression of IFNAR1.
Materials and Methods
Cell lines and viruses
MRC-5 (human embryonic lung fibroblast), RD (human rhabdomyosarcoma), and Vero E6 (African green monkey kidney) cells were maintained in Minimum Essential Medium (MEM; Hyclone) containing 10% fetal bovine serum (FBS; Hyclone) with 100 U/mL penicillin and 100 μg/mL streptomycin (Gibco). EV71 strains VR1432 (ATCC) and GDV103 (CCTCC), and vesicular stomatitis virus (VSV) were propagated in Vero E6 cells.
Plasmids
The sequences encoding 2A and 3C were amplified from GDV103 cDNA using the primers: 2A, 5′-CCC GAA TTC ATG GGG AAA TTT GGA C-3′ and 5′-CCC CTC GAG TTA CTT TCC ATG GCT T-3′; and 3C, 5′-CCC GAA TTC ATG GGC CCG AGC CTT G-3′ and 5′-CCC CTC GAG TTA CTT GTC ATC GTC GTC CTT GTA GTC TTG CTC ACT AGC AAA G-3′ and cloned into the vector pcDNA3.1 (+) (Invitrogen).
Cell transfection and stimulation
Transfection of HEK293T cells with the pcDNA3.1 (+) vectors expressing 2A or 3C was performed using TurboFect (Fermentas, LTU), according to the manufacturer's instructions.
EV71 titration
EV71 was diluted by serial 10-fold dilutions in MEM containing 2% FBS, and added to approximately 95% confluent Vero E6 cells cultured in 6-well plates. After adsorption at 37°C for 1 h, the viral supernatant was removed and replaced with 1.5% agarose overlay media composed of a 1:1 ratio of 2× MEM to 3% L.M.P. agarose (Sigma). After 96 h, the cells were fixed in 4% paraformaldehyde at room temperature for 3 h, the agarose overlay was carefully removed, the plaques were stained using crystal violet at 37°C for 30 min, and the number of plaques was counted after washing.
Reverse transcription polymerase chain reaction and quantitative real-time polymerase chain reaction
Total RNA was extracted from the cells using the RNAprep Pure Cell/Bacteria Kit (Tiangen) according to the manufacturer's instructions. In brief, the cells were lysed in the presence of β-mercaptoethanol and the lysates were loaded to a column, washed, and eluted in RNase-free water. Reverse transcription polymerase chain reaction (RT-PCR) was carried out using the First Strand cDNA Synthesis Kit (Fermentas), and the cDNA samples were subjected to PCR amplification and electrophoresis to detect IFN-β and β-actin. The primers used were as follows: human IFN-β, 5′-GAC CAA CAA GTG TCT CCT CCA AA-3′ and 5′-GAA CTG CTG CAG CTG CTT AAT C-3′; and β-actin, 5′-TCA CCA ACT GGG ACG ACA- 3′ and 5′-ACA GCC TGG ATA GCA ACG-3′.
Quantitative RT-PCR (qRT-PCR) was carried out using the ABI 7500A PCR system with Power SYBR Green Master Mix (Applied Biosystems) using the following primers: ISG56, 5′-CCT CCT TGG GTT CGT CTA CA-3′ and 5′-TTC TCA AAG TCA GCA GCC AGT-3′; MxA, 5′-ATA GAC CTT CCT GGC ATA ACC A-3′ and 5′-CTT CAG TTC CTT TGT CCA CCA-3′; OAS1, 5′-CTA CCC TGT GTG TGT GTC CAA G-3′ and 5′-CAC GGA AAA TGC TCT CTC TCT T-3′, and JAK1, 5′-TTT ACC ATC AAT CAT CAC CCT GT-3′ and 5′-CTA CCT CCA AGC AAA CTG AAA ACT-3′. The relative amount of each target mRNA was normalized to the level of β-actin in each individual sample.
Western blotting
Cells were lysed in 1× SDS sample buffer (62.5 mmol/L Tris-HCl, pH 6.8, 2% SDS [w/v], 10% glycerol, 50 mmol/L DTT, 0.01% bromophenol blue [w/v], and 1× protease inhibitor cocktail [Thermo Scientific]) on ice, sonicated for 10–15 sec, heated at 95°C for 5 min, and the supernatants were collected by centrifugation at 12,000 g for 5 min. The same amount of total protein from each sample was fractionated by 12% SDS-PAGE and transferred to nitrocellulose membranes (GE Healthcare). The membranes were blocked with 5% skimmed milk in TBST (20 mmol/L Tris-HCl, pH 7.4, 150 mmol/L NaCl, 0.1% Tween 20) for 1 h, and then incubated with specific antibodies according to the manufacturer's instructions. The unphosphorylated forms of STAT1 and STAT2 were detected using anti-STAT1 and anti-STAT2 antibodies (sc-464 and sc-1668; Santa Cruz Biotechnology). The phosphorylated forms of STAT1 and STAT2 were detected using anti-phospho-STAT1 (#9167; Cell Signaling Technology) and anti-phospho-STAT2 (sc-21689-R; Santa Cruz Biotechnology). The unphosphorylated forms of JAK1 and TYK2 were detected using anti-JAK1 and anti-TYK2 antibodies (sc-136225 and sc-5271; Santa Cruz Biotechnology). An EV71-specific antibody (ab36367; Abcam) was used for detection of viral proteins. IFNAR1 was detected using specific antibodies against IFNAR1 (sc-7391; Santa Cruz Biotechnology). All secondary antibodies were purchased from Santa Cruz Biotechnology. Each immunoblot was performed at least three times.
Flow cytometry
Cells were gently removed using cell dissociation buffer (GIBCO), centrifuged at 500 g for 5 min at 4°C, washed twice with ice-cold wash solution (PBS containing 2% FBS), incubated with mouse anti-IFNAR1 (sc-7391; Santa Cruz Biotechnology) for 30 min, followed by incubation with FITC-conjugated goat anti-mouse secondary antibody (sc-2010; Santa Cruz Biotechnology) for 30 min in the dark. The cells were washed, fixed in 2% paraformaldehyde, and the surface expression of IFNAR1 was analyzed by flow cytometry using a FACSCalibur (BD) equipped with CELLQuest software.
Statistical analysis
Data are expressed as mean±standard error of the mean (SEM) and were analyzed using one-way or two-way analysis of variance (AVOVA) with Prism 5 (GraphPad Software).
Results
EV71 infection slightly induces the expression of IFN-β
To explore the effect of EV71 on the cellular response to type I IFN, we examined IFN-β mRNA at different time points postinfection in cells infected with EV71. MRC-5 cells were mock infected or infected with EV71 strain GDV103 or strain VR1432 at a multiplicity of infection (MOI) of 10. Vesicular stomatitis virus (VSV) was used as a positive control to induce IFN-β expression. RT-PCR and qRT-PCR were conducted to detect the expression of IFN-β at 0, 4, 8, and 12 h postinfection (hpi). As the RT-PCR results show in Figure 1A, VSV stimulated the expression of IFN-β in MRC-5 cells, whereas EV71 failed throughout infection. qRT- PCR results show that EV71 infection could increase the expression of IFN-β by 1–3-fold compared to that in mock-infected cells(Fig. 1B and C). Together, these results suggest that EV71 is not a potent IFN-β inducer.
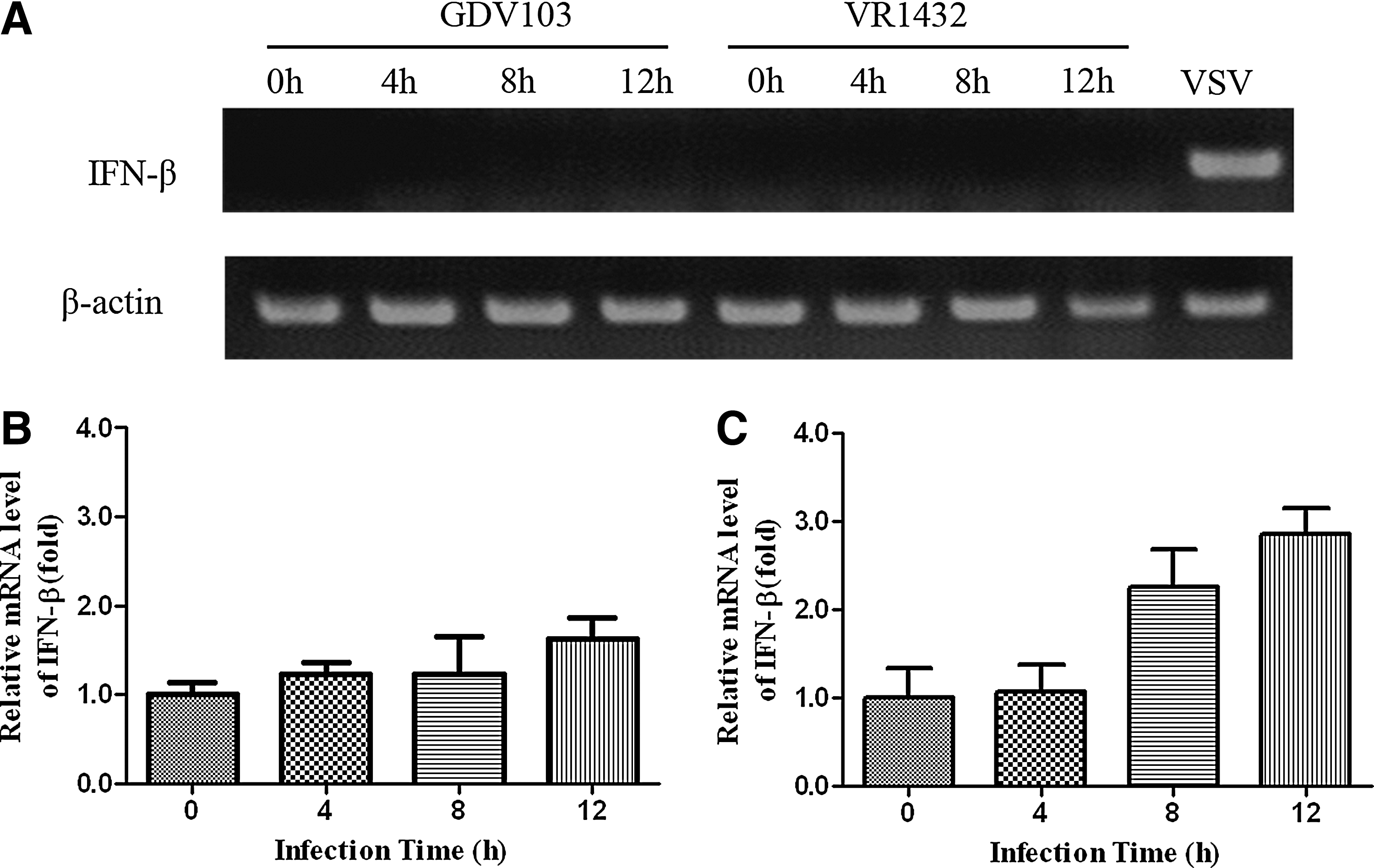
Enterovirus 71 (EV71) infection slightly activated cellular interferon (IFN)-β mRNA. MRC-5 cells were mock infected or infected with EV71 strain GDV103 or strain VR1432 at a multiplicity of infection (MOI) of 10 for the indicated times. As a control, mock-infected cells were infected with vesicular stomatitis virus (VSV).
EV71 interferes with the expression of IFN-stimulated genes
To investigate the mechanism by which EV71 inhibits type I IFN signaling, we quantified the expression of three IFN-stimulated genes (ISGs): ISG56, MxA, and OAS1. MRC-5 cells were mock infected or infected with EV71 strain GDV103 at a MOI of 10 for 10 h, untreated or stimulated with IFN-α2b (1000 IU/mL) for 2 h, and total RNA was extracted to measure the expression of the three ISGs by qRT-PCR. EV71 strain GDV103 did not induce expression of any of the ISGs. However, IFN-α2b increased the expression of ISG56, MxA, and OAS1 by 29.7-, 36.4-, and 65.9-fold compared to mock-infected cells (Fig. 2). The expression levels of ISG56, MxA, and OAS1 were ∼4-, ∼8.1- and ∼9.9-fold lower, respectively, in GDV103-infected MRC-5 cells treated with IFN-α2b than that in mock MRC-5 cells treated with IFN-α2b. These results indicate that EV71 antagonizes type I IFN signaling by inhibiting the expression of ISGs.
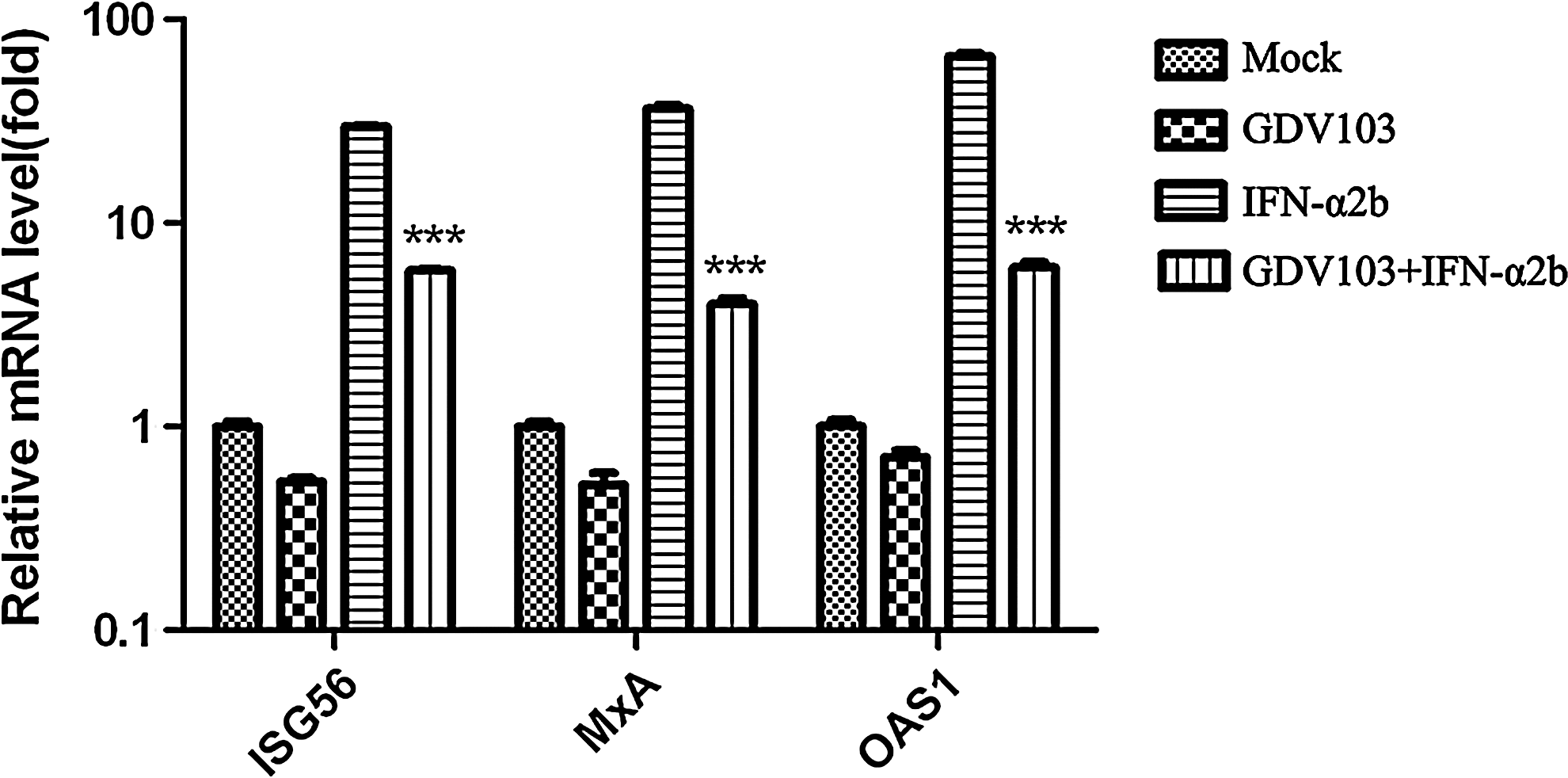
EV71 infection inhibits the cellular expression of type I IFN-stimulated genes. MRC-5 cells were mock infected or infected with EV71 strain GDV103 at a MOI of 10 for 10 h, and then treated with IFN-α2b for 2 h. Total RNA was extracted and used to measure the expression levels of ISGs by qRT-PCR; ***p<0.001 versus type I IFN.
EV71 infection inhibits IFN-α2b-induced STAT1 and STAT2 phosphorylation
STAT1 and STAT2 play a key role in the type I IFN-activated JAK-STAT signaling pathway (4,27,33). A number of viruses have been reported to inhibit type I IFN-induced JAK-STAT signaling by inhibiting STAT1 and STAT2 phosphorylation (5,612,20). Therefore, we analyzed whether the phosphorylation of STAT1 and STAT2 in response to IFN-α2b were altered in EV71-infected cells. MRC-5 and RD cells were infected with EV71 strain GDV103 or strain VR1432 at a MOI of 10 for different periods of time, and then untreated or stimulated with 1,000 IU/mL IFN-α2b for 1 h. As shown in Figure 3, neither EV71 strain GDV103 or strain VR1432 induced the phosphorylation of STAT1 or STAT2 in MRC-5 or RD cells in the absence of IFN-α2b treatment. However, the phosphorylation of STAT1 and STAT2 were markedly increased by IFN-α2b treatment in MRC-5 or RD cells infected with EV71 strain GDV103 or strain VR1432 at 0 and 6 hpi (Fig. 3). However, EV71 protein expression could not be detected in MRC-5 or RD cells infected with EV71 strain GDV103 or strain VR1432 until 9 hpi, and the levels of phosphorylated STAT1 and STAT2 significantly reduced in IFN-α2b-treated infected cells at 12 hpi. These results indicate that EV71 infection inhibits the IFN-α2b-induced phosphorylation of STAT1 and STAT2 in both MRC-5 and RD cells.
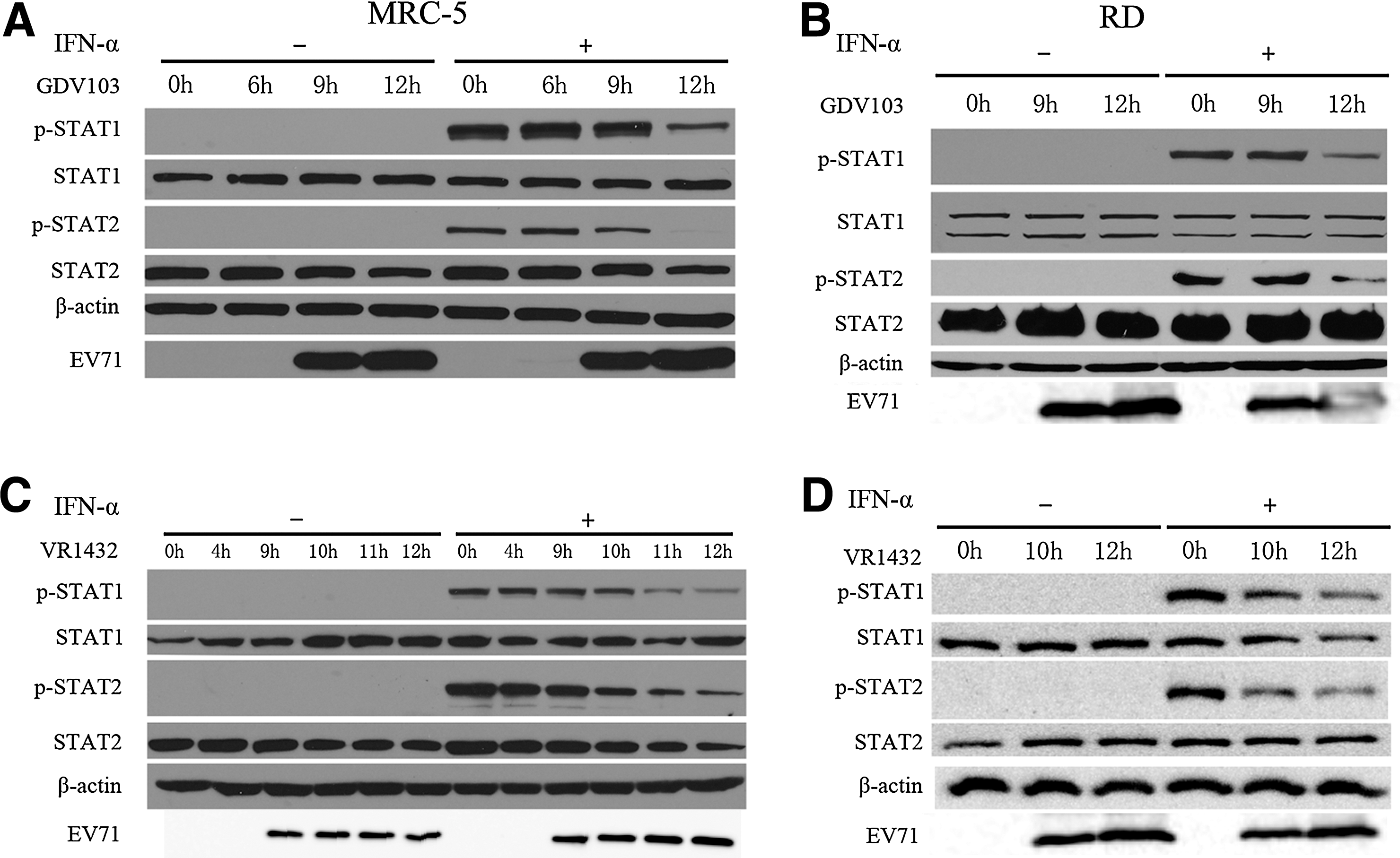
EV71 infection inhibits cellular STAT1 and STAT2 phosphorylation. MRC-5 or RD cells were infected with EV71 strain GDV103
EV71 infection does not alter cellular surface expression of the IFNAR1 subunit
Some viruses can disrupt JAK-STAT signaling by reducing the cellular surface expression of the interferon receptors (3,8,32,36), and Lu et al. previously reported that EV71 disrupted IFN signaling by reducing the expression of IFNAR1 at 6 hpi (18). As EV71 infection inhibited the phosphorylation of STAT1 and STAT2, we investigated whether the total expression level and surface expression of IFNAR were affected in EV71-infected cells. MRC-5 cells were infected with EV71 strain GDV103 or strain VR1432 at a MOI of 10 for different periods of time, then the cells were untreated or treated with IFN-α2b (1,000 IU/mL) for 1 h, and the cell lysates were collected to assess the total protein expression levels and surface expression of IFNAR1 using an IFNAR1-specific antibody by Western blotting and FACS analysis. However, neither GDV103 (Fig. 4A and 4E) nor VR1432 (Fig. 4B and 4F) infection significantly altered the total protein expression level of IFNAR1 in MRC-5 cells. Treatment with IFN-α2b (1,000 IU/mL) did not reduce the number of GDV103 or VR1432-infected MRC-5 cells expressing IFNAR1 on the cell surface (Fig. 4C and D).
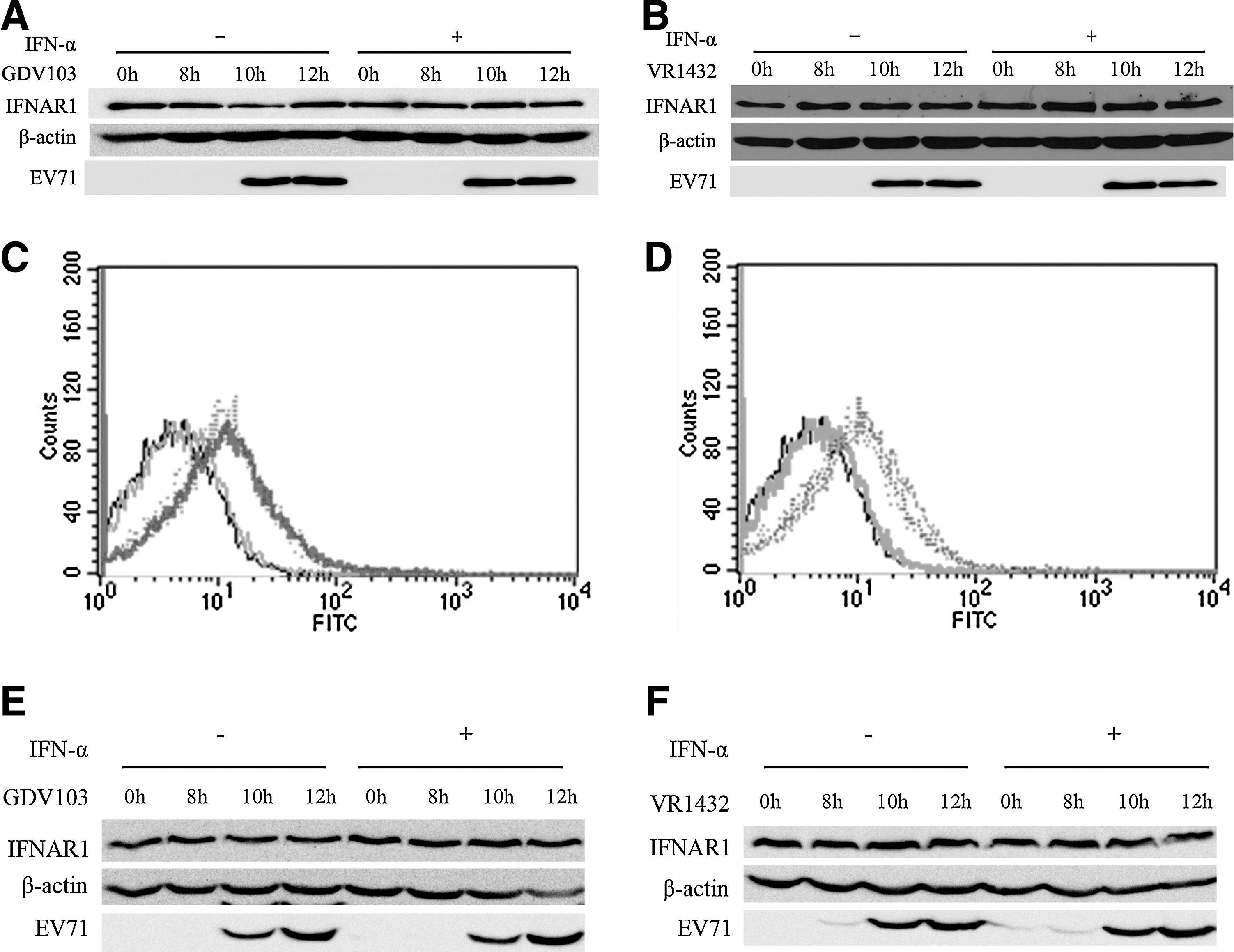
EV71 infection does not affect cellular surface expression of the IFNAR1 subunit.
Impaired phosphorylation of STAT1 and STAT2 in EV71-infected cells is not mediated by the protein tyrosine phosphatases
Protein tyrosine phosphatases (PTPs) are one of the key cellular regulators that negatively regulate JAK-STAT signaling. The PTPs can directly dephosphorylate STAT1 and STAT2, or act indirectly by dephosphorylating and inactivating JAKs resulting in reduced phosphorylation of STAT1 and STAT2 (20). To determine if PTPs play a role in the inhibition of type I IFN signaling by EV71, we examined whether pretreatment of the cells with sodium vanadate, a PTP inhibitor, could reverse the inhibitory effect of EV71 on JAK-STAT signaling (11). If the inhibition of IFN-α-induced STAT1 and STAT2 phosphorylation is mediated by the PTPs, then sodium vanadate should rescue the expression of p-STAT1 and p-STAT2 in EV71-infected cells. Mock-infected cells and cells infected with EV71 strain GDV103 for 9 or 12 h were incubated with 25 μM sodium vanadate for 15 min, and then stimulated with IFN-α2b (1,000 IU/mL) for 1 h. As shown in Figure 5, inhibition of the PTPs using sodium vanadate did not rescue the EV71-induced suppression of STAT1 and STAT2 phosphorylation.
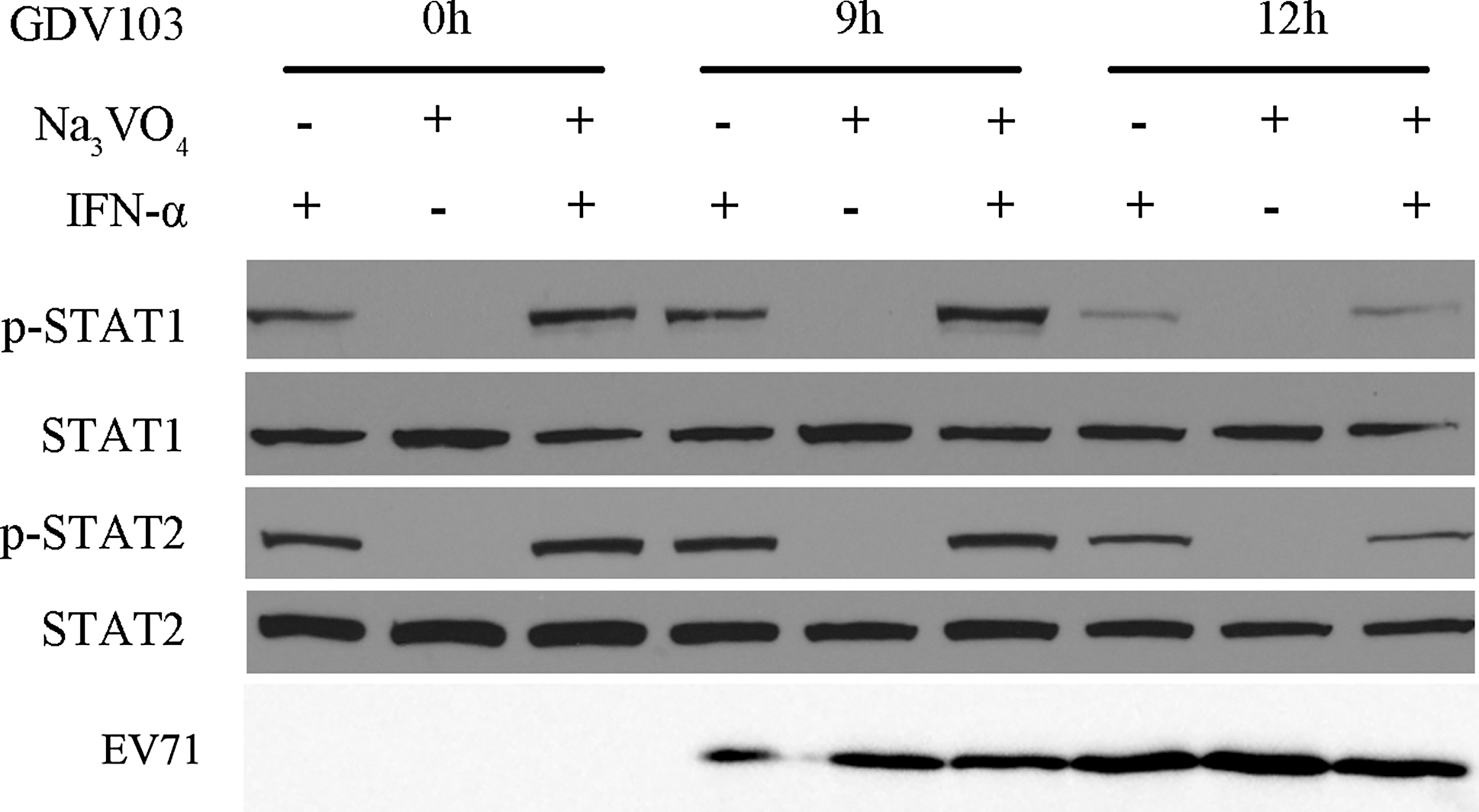
Inhibition of protein tyrosine phosphatases (PTPs) using sodium vanadate does not reverse the inhibition of IFN-stimulated STAT1 and STAT2 phosphorylation in EV71-infected cells. MRC-5 cells were infected with EV71 strain GDV103 at a MOI of 10 for the indicated times, then treated with or without sodium vanadate for 15 min prior to stimulation with IFN-α2b (1,000 IU/mL) for 1 h. Cells were harvested, and the protein lysates were subjected to Western blot analyses using anti-phospho-STAT1, anti-STAT1, anti-phospho-STAT2, and anti-STAT2 antibodies; β-actin was detected as a loading control.
EV71 infection does not affect the protein expression levels of SOCS1 or SOCS3
In addition to the PTPs, phosphorylation of STAT1 and STAT2 can also be regulated by other cellular regulators such as the suppressor of cytokine signaling (SOCS) family. To investigate whether EV71 affected the expression of SOCS1 and SOCS3, MRC-5 cells were infected with EV71 strain GDV103 for different periods of time, and then stimulated with IFN-α2b (1,000 IU/mL) for 1 h. As shown in Figure 6, the protein expression levels of SOCS1 and SOCS3 were not significantly altered in EV71-infected cells, while the levels of phosphorylated STAT1 reduced remarkably.
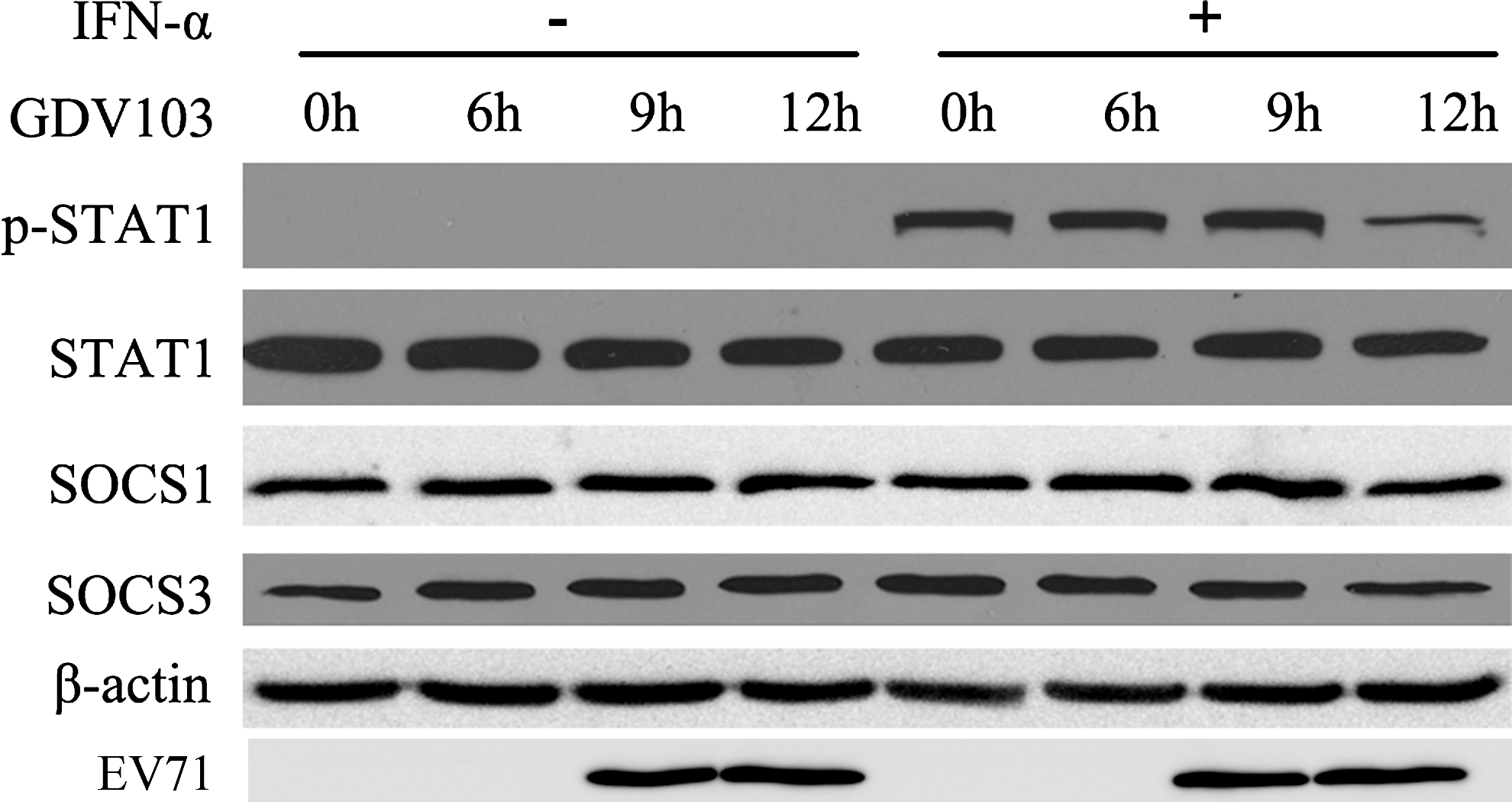
EV71 infection does not affect the cellular expression of SOCS1 and SOCS3. MRC-5 cells were infected with EV71 strain GDV103 at a MOI of 10 for 6, 9, or 12 h, and then stimulated with IFN-α2b (1,000 IU/mL) for 1 h. Cells were harvested and the protein lysates were subjected to Western blot analyses using STAT1, p-STAT1, SOCS1, and SOCS3 antibodies; β-actin was detected as a loading control.
EV71 infection blocks the phosphorylation of both JAK1 and TYK2 by downregulating JAK1 but not TYK2
The first step in activation of the type I IFN signal transduction cascade is autophosphorylation of JAK1 and TYK2 in response to ligand-induced conformational changes in the IFN-α/β receptors (12). Therefore, we examined the expression and activation of the JAK kinases in EV71-infected MRC-5 and RD cells. Infection with EV71 strain GDV103 (Fig. 7A and B) or strain VR1432 (Fig. 7C and D) markedly downregulated the expression of JAK1 at 10 hpi, and correlated with a loss of detectable phosphorylated JAK1 and TYK2. However, infection with EV71 strain GDV103 or strain VR1432 did not significantly affect the expression of TYK2 (Fig. 7).
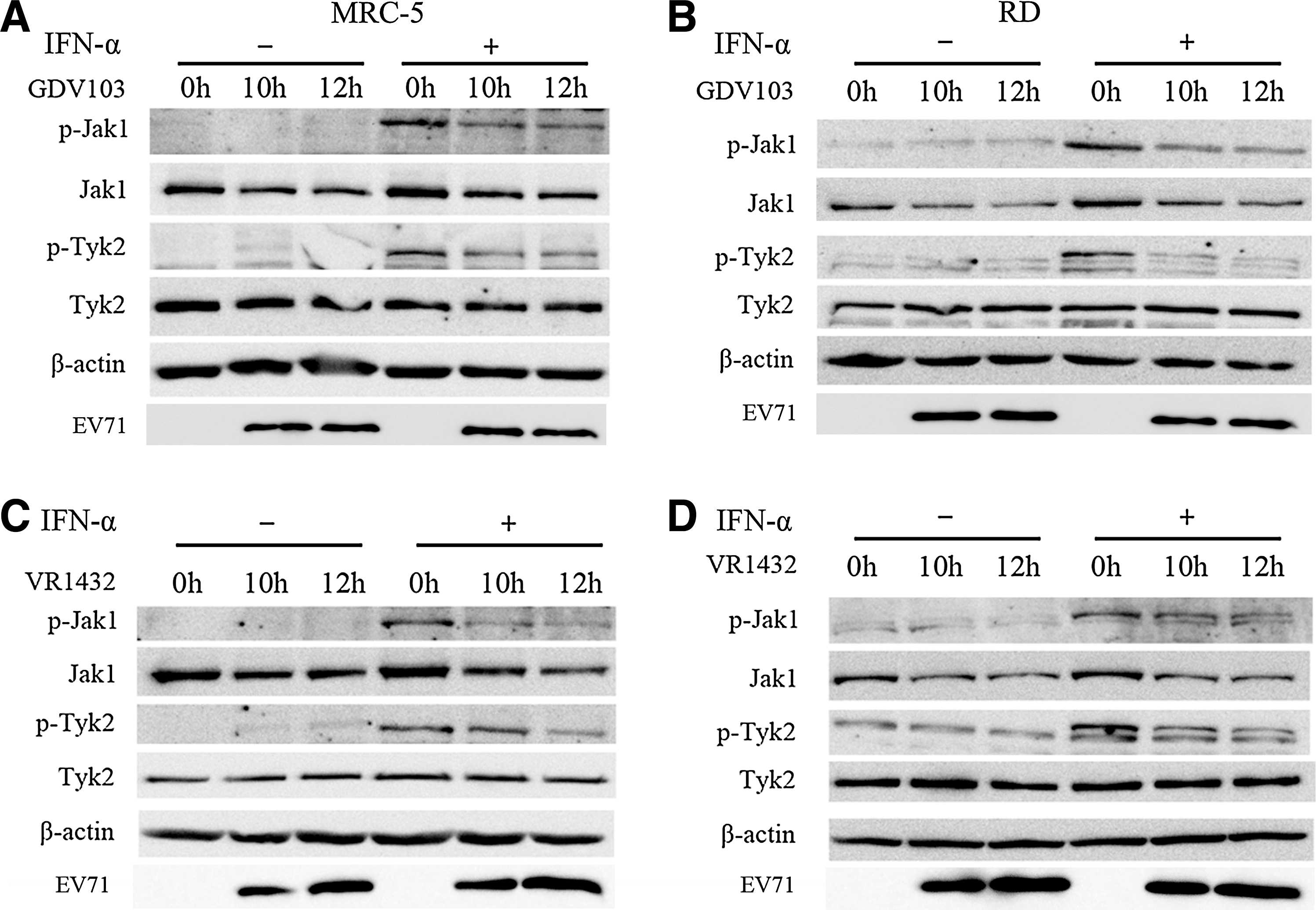
EV71 infection downregulates cellular JAK1 protein expression. MRC-5 cells or RD cells were infected with EV71 strain GDV103
EV71 infection does not decrease JAK1 mRNA expression but leads to proteasome-independent degradation of JAK1
Next, we quantified the expression of JAK1 mRNA in MRC-5 cells infected with EV71 strain GDV103 (Fig. 8A) or strain VR1432 (Fig. 8B) at 0, 8, 10, and 12hpi. As shown in Figure 8, the expression of JAK1 mRNA did not decrease in EV71-infected cells, which suggests that the downregulation of JAK1 in EV71-infected cells occurs at a post-transcriptional level.
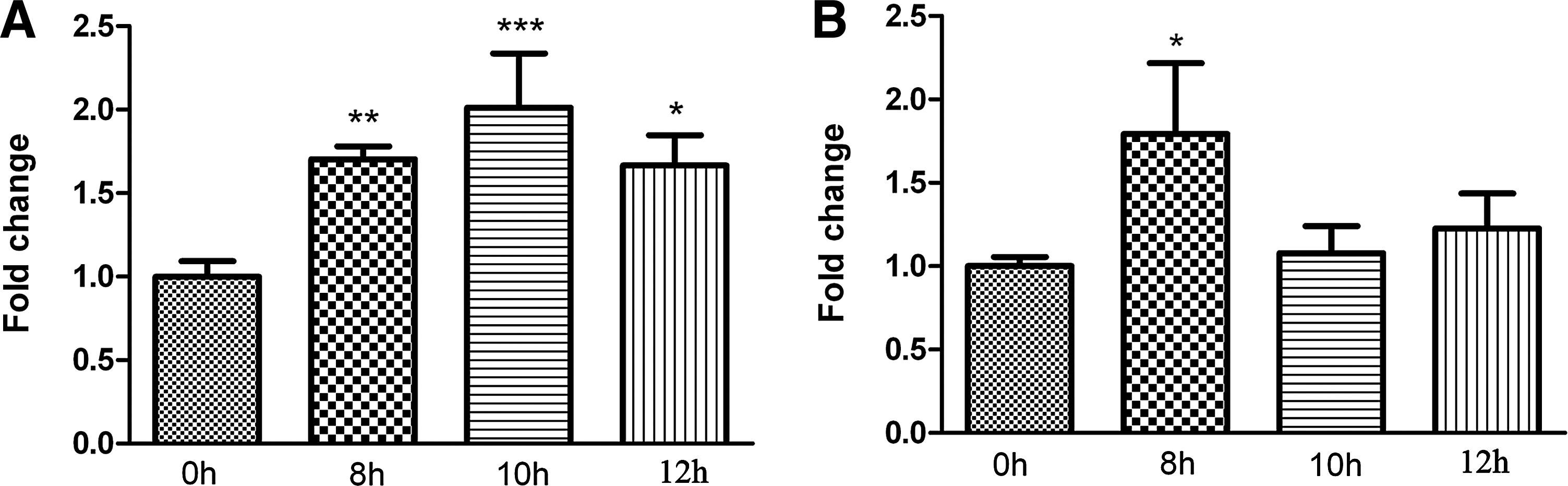
EV71 infection does not downregulate cellular JAK1 mRNA expression. Total RNA was extracted from MRC-5 cells infected with EV71 strain GDV103
Proteasome-dependent degradation is a cellular mechanism commonly adopted by viruses to modulate the expression of signaling proteins. For example, human parainfluenza virus type 2 and respiratory syncytial virus (RSV) both induce the downregulation of STAT2 via a proteasome-dependent pathway (1,23), and human metapneumovirus and herpes simplex virus 1 both inhibit JAK-STAT signaling by decreasing the expression of JAK1 (3,25). To investigate whether EV71 induces downregulation of JAK1 via a proteasome-dependent pathway, MRC-5 cells were pretreated with the cell-permeable, nonspecific proteasome inhibitor MG-132, then mock infected or infected with EV71 strain GDV103 (Fig. 9A) or strain VR1432 (Fig. 9B) at a MOI of 10 for 12 h, and harvested to measure JAK1 protein expression by Western blotting. MG132 did not clearly alter the effects of EV71 strain GDV103 or strain VR1432 on the protein expression level of JAK1, as the range of JAK1 expressed in mock-infected and EV71-infected cells was similar whether MG-132 was added or not (Fig. 9). These results indicate that JAK1 is not downregulated via a proteasome-dependent pathway in EV71-infected cells, which suggests that EV71 leads to downregulation of JAK1 via a proteasome-independent mechanism.
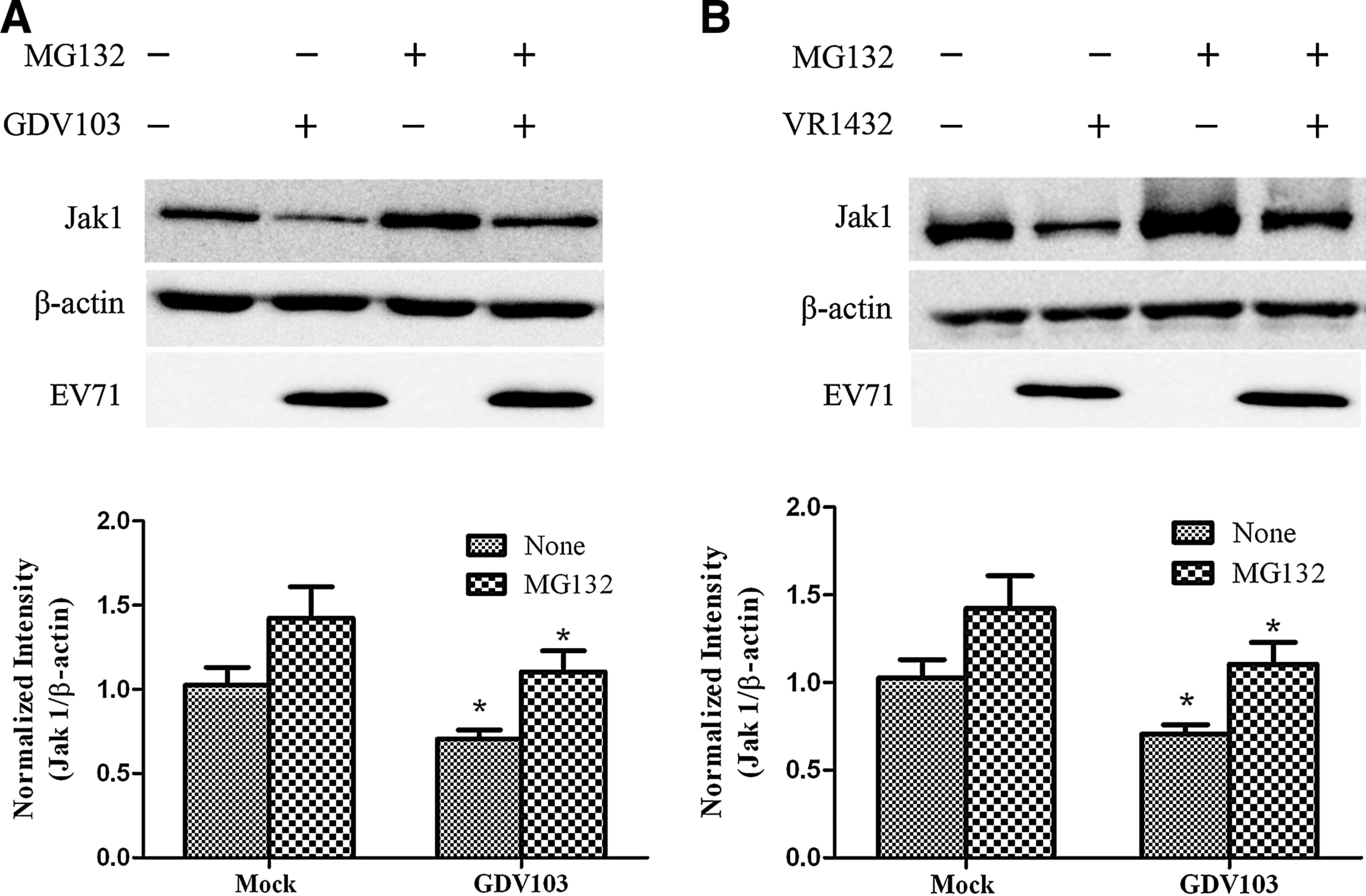
EV71-induced downregulation of cellular JAK1 occurs via a proteasome-independent mechanism. MRC-5 cells were pretreated with 5 μmol/L MG132 for 1 h, then mock-infected or infected with EV71 strain GDV103
The viral nonstructural proteins 2A and 3C are not antagonists of type I IFN signaling in EV71-infected cells
Due to their protease activity, the nonstructural proteins 2A and 3C have been considered as the key viral proteins that antagonize cellular type I IFN signaling. Since we observed that EV71 strain GDV103 inhibited IFN-α signaling by decreasing the expression of JAK1 and subsequently blocking the phosphorylation of STAT1 and STAT2 (Figs. 3 and 7), we next examined whether the phosphorylation of STAT1 and STAT2 were suppressed by the viral proteins 2A and 3C. To investigate the potential antagonistic activity of 2A and 3C toward type I IFN signaling, we generated plasmids to express the viral proteins 2A and 3C encoded by GDV103. HEK293T cells were transiently transfected with plasmids expressing the viral proteins 2A or 3C, and 24 h after transfection, the cells were stimulated with or without IFN-α2b for 1 h. Cleavage of eIF4G represents the functional expression of 2A, as it was hard to detect directly due to its inhibition on its own translation (15); and an antiflag antibody was used to detect the expression of flag-tagged 3C in transfected cells. As shown in Figure 10, JAK1 expression did not decrease, and IFN-α-stimulated STAT1 and STAT2 phosphorylation were not blocked in HEK293T cells overexpressing the viral protein 2A or 3C.
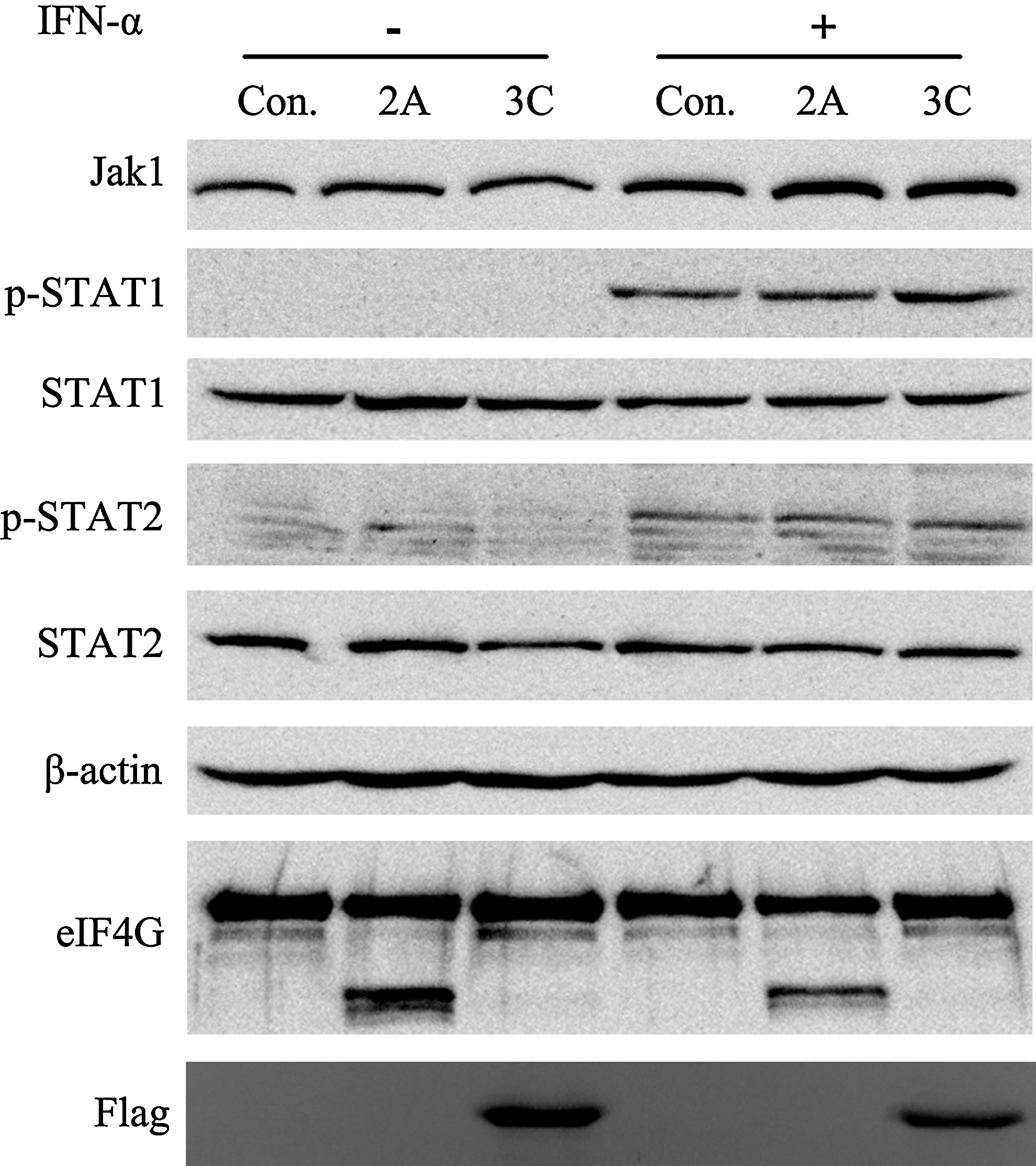
Expression of the EV71 viral proteins 2A and 3C does not inhibit cellular phosphorylation of STAT1 and STAT2. HEK 293T cells were transfected with a control vector or plasmids expressing the viral proteins 2A or 3C; 24 h after transfection, the cells were treated with or without 1,000 IU/mL IFN-α2b for 1 h. Cells were harvested, and the protein lysates were subjected to Western blot analyses using antibodies against STAT1, STAT2, p-STAT1, p-STAT2, eIF4G, and Flag; β-actin was detected as a loading control.
Discussion
Type I IFN can be rapidly activated in most cell types, mediates a wide variety of antiviral responses, and represents an important initial barrier to viral infection (25). To combat host antiviral defenses, viruses have developed a diverse range of strategies to circumvent type I IFN responses, which can be grouped into two main mechanisms: inhibition of type I IFN production by inactivating the transcription factors through targeting adaptor in the initiation step involved in the expression of type I IFN, such as IRF3/IRF7, NFκB, or ATF-2/c-jun; or inhibition of type I IFN-induced activation of ISGs via the JAK-STAT signaling pathway (35,37). This study demonstrates that EV71 facilitates viral invasion and reduces the cellular antiviral response by inhibiting JAK-STAT signaling.
Functional EV71 precursor proteins and proteins are produced upon maturation and cleavage of viral polyproteins by the virus-encoded proteases 2Apro and 3Cpro (39). Recently, some picornaviruses have been reported to impair downstream cellular type I IFN production by disrupting the toll-like receptors (TLRs) or retinoic acid inducible gene-1 (RIG1)/melanoma differentiation associated gene-5 (MDA5) receptor pathway. For example, the hepatitis A virus 3CD protease-polymerase (21), 3C protein of EV71 and foot-and-mouth disease virus (FMDV) (16) use their protease activity to cleave the adaptor TRIF (TIR domain-containing adaptor) or modulator NEMO (NF-κB essential modulator), which are required for the activation of NFκB and IRF3. As a result, the transcription of type I IFN-β can be detected slightly in EV71-infected cells (Fig. 1).
Viral proteins can disrupt every stage of the type I IFN-induced JAK-STAT signaling pathway in order to enable successful invasion and replication in host cells. The phosphorylation of STAT1 and STAT2 are regarded as hallmarks of the activation of type I IFN signaling (18). Our results showed that infection with EV71 significantly inhibited the phosphorylation of STAT1 and STAT2 (Fig. 3). The first step in the activation of ISG induction via the JAK-STAT signaling pathway is the binding of type I IFN to IFNAR. It has been reported that EV71 disrupts type I IFN signaling by downregulating IFNAR1, and the viral 2A protease encoded by EV71 has been proven to be an antagonist of type I IFN (18). However, our data show that EV71 infection did not alter the total expression level or surface expression of IFNAR1 (Fig. 4). In our study, EV71 (GDV103 and VR1432) infection induced IFN-β slightly and did not reduce the expression of IFNAR1 (Western blotting and FACS analysis showed the same result), which differs from Lu's publication. This could be because of different strains of the EV71 virus, the different cell lines (even the same cell line is from different organization), and a different lab. In our work, we used the same cell line (RD cell) and a different cell line (MRC-5 cell), but we did not get the same results as in Lu's study.
In our study, EV71 infection downregulated JAK1 protein expression but did not significantly affect the expression of TYK2. JAK1 is a member of the Janus family of protein tyrosine kinases, which are characterized by the presence of a second phosphotransferase-related or pseudokinase domain near the functional kinase domain (24). JAK1 is essential for the phosphorylation and activation of TYK2 in the type I IFN receptor complex (10). Furthermore, the mRNA levels of JAK1 did not decrease after EV71 infection as the protein expression. As proteasome-dependent degradation of signaling proteins is a common mechanism for viral modulation of cellular signaling proteins (22,23,42), the proteasome inhibitor MG132 was used to investigate if JAK1 was downregulated via a proteasome-dependent mechanism. However, we observed that downregulation of JAK1 in EV71-infected cells did not occur via a proteasome-dependent mechanism. Additionally, several endogenous cellular proteins have been identified to act as negative regulators of the JAK-STAT pathway, including SOCS, the PTPs, and protein inhibitor of activated STATs (PIAS) (29). Of these, SOCS1 and SOCS3 are induced by type I IFN and can directly interact with and inhibit JAK function via a negative feedback loop (7,26,34,41). The human T-cell leukemia virus type 1 and influenza A virus have been reported to inhibit type I IFN antiviral signaling via induction of SOCS1 and SOCS3 respectively (2,20). Our data reveal that the inhibition of type I IFN-induced phosphorylation of the JAKs and STATs in EV71-infected cells was not mediated by the PTPs, SOCS1, or SOCS3 (Figs. 5 and 6). We also demonstrate that the viral proteins 2A and 3C of EV71 did not act as antagonists to inhibit the phosphorylation of STAT1 and STAT2. The other nonstructural viral proteins, including 2B, 2C, 3A, 3B, and 3D, have no significant effect on phosphorylation of STAT1 and STAT2 either (data not shown). Perhaps the interactions of two or more viral proteins may inhibit the IFN signaling. Taken together, these results indicate that EV71 may downregulate JAK1 by inhibiting the nucleo-cytoplasmic transport or translation of cellular JAK1 mRNA, or via some other proteasome-independent degradation or autophagy-lysosomal pathway. Given the prominent role of JAK1 in numerous cellular signaling pathways, the impact of EV71-mediated downregulation of JAK1 on type I IFN signaling requires further investigation.
Conclusions
This study demonstrates that infection with the EV71 virus inhibits cellular type I IFN-induced signaling by downregulating the expression of JAK1, rather than IFNAR1. The EV71-induced downregulation of JAK1 occurs at a post-transcriptional level via a proteasome-independent mechanism, and induces inhibition of JAK1 and TYK2 phosphorylation, in parallel to the impaired phosphorylation of STAT1 and STAT2. Though the detailed mechanism of JAK1 downregulaiton remains unclear, we ruled out the antagonists roles of EV71 viral proteins 2A and 3C. This study supplies another mechanisms by which EV71 inhibits the cellular type I IFN response, which could provide us with new understanding and potential antiviral targets of EV71 infection.
Footnotes
Acknowledgments
We would like to thank Dr. Ruiyuan Cao for his kind help with viral culture. We would also like to thank Prof. Hui Zhong for his valuable suggestions. This work was supported by the National Natural Science Foundation of China (81025018).
Author Disclosure Statement
No competing financial interests exist.