
Abstract
Antiretroviral therapy (ART) represents a significant milestone in the battle against AIDS. However, we continue learning about HIV and confronting challenges 30 years after its discovery. HIV has cleverly tricked both the host immune system and ART. First, the many HIV subtypes and recombinant forms have different susceptibilities to antiretroviral drugs, which may represent an issue in countries where ART is just being introduced. Second, even under the suppressive pressures of ART, HIV still increases inflammatory mediators, deregulates apoptosis and proliferation, and induces oxidative stress in the host. Third, the preference of HIV for CXCR4 as a co-receptor may also have noxious outcomes, including potential malignancies. Furthermore, HIV still replicates cryptically in anatomical reservoirs, including the lung. HIV impairs bronchoalveolar T-lymphocyte and macrophage immune responses, rendering the lung susceptible to comorbidities. In addition, HIV-infected individuals are significantly more susceptible to long-term HIV-associated complications. This review focuses on chronic obstructive pulmonary disease (COPD), pulmonary arterial hypertension, and lung cancer. Almost two decades after the advent of highly active ART, we now know that HIV-infected individuals on ART live as long as the uninfected population. Fortunately, its availability is rapidly increasing in low- and middle-income countries. Nevertheless, ART is not risk-free: the developed world is facing issues with antiretroviral drug toxicity, resistance, and drug–drug interactions, while developing countries are confronting issues with immune reconstitution inflammatory syndrome. Several aspects of the complexity of HIV persistence and challenges with ART are discussed, as well as suggestions for new avenues of research.
Introduction
T
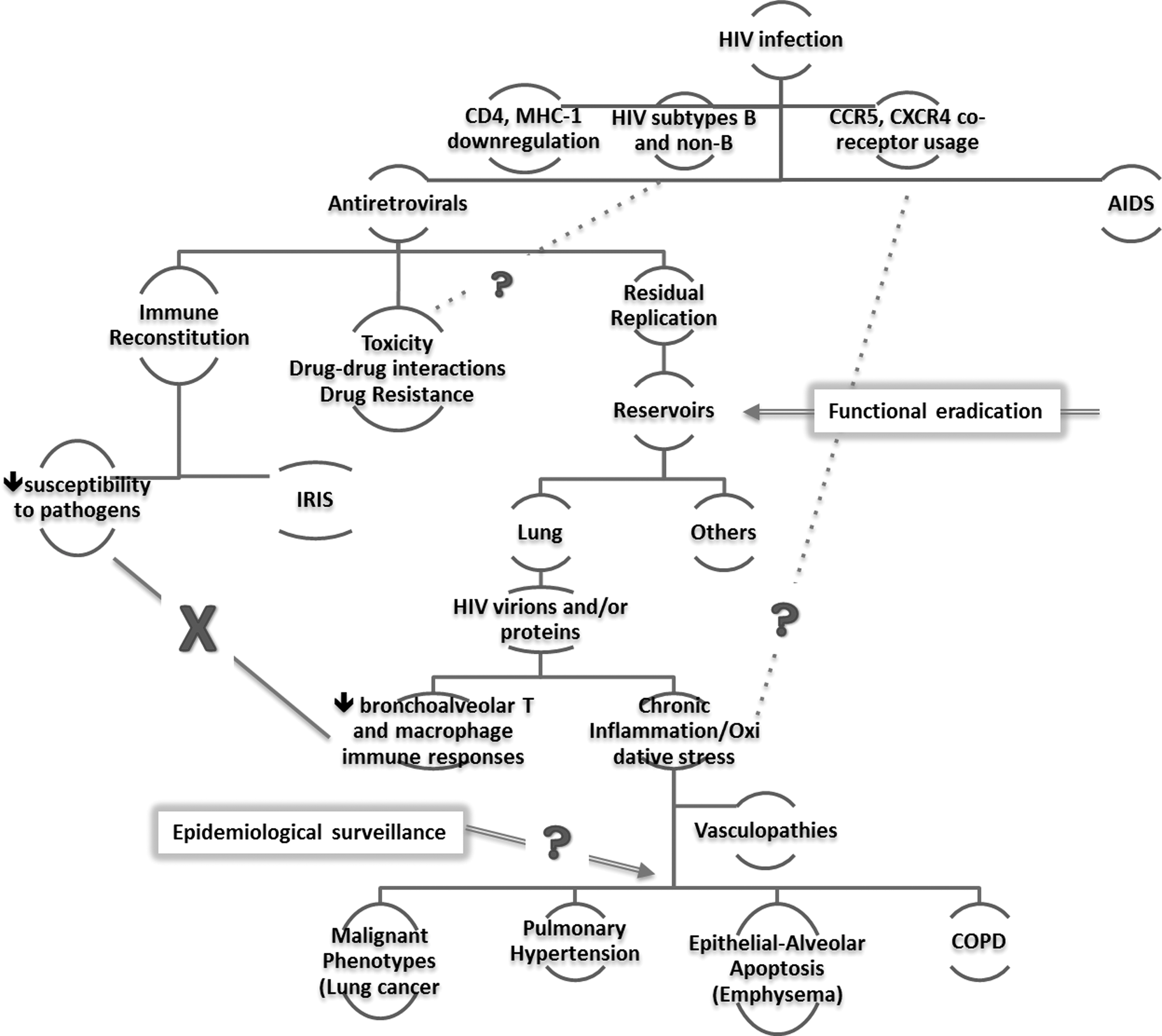
Interrelationship between concepts of HIV persistence and pathogenesis and challenges in the era of antiretroviral therapy. The outcomes of HIV infection may be influenced by downregulation of critical host cell receptors (i.e., CD4 and MHC-1), the subtype of the infecting virion and the co-receptor usage. The advent of highly active antiretrovirals has brought an end to AIDS. Antiretrovirals decrease HIV viral loads to levels below the limits of clinical detection and restores the overall immunity. While the reconstituted immune system protects the host from opportunistic pathogens, the vigorous response to residual pathogens (i.e., IRIS) is concerning low- and middle-income countries where antiretrovirals are increasingly available. However, long-term use of antiretrovirals causes several side effects; additional issues include drug–drug interactions and drug resistance. Even under suppressive pressures of antiretrovirals, HIV still replicates at low levels and evolves in anatomical reservoirs; efforts are aimed at the functional eradication of these reservoirs. HIV is present in the lung, where the HIV virions and/or proteins compromise the innate immunity and induce oxidative stress and chronic inflammation, leading to vasculopathies. People living with HIV are significantly more susceptible to pulmonary complications, including lung cancer, pulmonary hypertension, and COPD. MHC-1, major histocompatibility complex-1; IRIS, immune reconstitution inflammatory syndrome; COPD, chronic obstructive pulmonary disease.
HIV: The Portrait of a Challenging Virus
Co-discovered by Barre-Sinoussi et al. and Gallo et al. three decades ago (8,55), HIV is the etiologic agent of AIDS and the topic of more than 270,000 research articles in the biomedical literature. HIV produces more than 10 billion virions per day and has an extensive genetic variability, compelled by the error-prone viral transcription (139,156), rapid HIV turnover (new generation every 2.6 days) (130), genetic recombination (126), and host selective immune pressures. Due to such genetic variability, the main group of HIV type 1 is subtyped into nine genetic variants (A, B, C, D, F, G, H, J, and K), all of which recombine, which may introduce differences in mutation rates and fitness. HIV mutation rates do not seem to vary among host cell types, at least in embryonic kidney cell lines, T-lymphoblasts, and glioblastoma (77). Whether this holds true in primary cells, especially in tissue-resident cell types with different metabolic demands, remains to be addressed.
Understanding HIV interactions with the host is essential to learn how the viral machinery induces pathogenicity and to identify potential therapeutic targets. Key concepts on HIV entry, persistence, and pathogenesis are discussed.
HIV receptors
HIV enters the cells via interactions with the CD4 receptor in the host cell and the C-C chemokine receptor-5 (CCR5) and C-X-C chemokine receptor-4 (CXCR4). The CCR5 is a receptor for RANTES/CCL5, MIP-α/CCL3, and MIP-β/CCL4 in primary macrophages (42). The CCR5 receptor is expressed in microglia, T-lymphocytes, macrophages, and dendritic cells (DC). CXCR4, initially known as “fusin,” is a 7-transmembrane G protein-coupled receptor used by HIV as a co-receptor for preferential entry to T-cell lines (53). Its natural ligand is stromal derived factor-1 (SDF-1/CXCL12) (10). Conventionally, HIV virions that use CCR5 as a portal of entry are designated as “R5,” while virions using CXCR4 are referred to as “X4.” The HIV preference for CCR5 co-receptor switches to a preference for CXCR4 over the course of HIV infection; this co-receptor switch predicts progression to AIDS in ∼50% of HIV+ individuals (29).
HIV-mediated evasion of immune surveillance
Shotgun proteomic analyses of the bronchoalveolar lavage fluid (BALf ) in HIV+ versus HIV− patients confirmed down-modulation of innate and adaptive immune responses such as lymphocyte-mediated immunity, activation of complement, and humoral responses (122). Furthermore, HIV may hide in cells by downregulation of key host receptors to evade immune surveillance (67). For example, HIV-Nef is a key player in HIV pathogenesis by enhancing infectivity and downregulating critical molecules such as major histocompatibility complex-1 (MHC-1) and CD4 receptors (115). It is known that Nef downregulates the CD4 receptor by targeting it to the endocytic degradation pathway in clathrin-coated vesicles. CD4 downregulation stimulates viral replication in primary T-cells (107). HIV-Nef also downregulates MHC-1 by sequestering it in the trans-Golgi, and hence it prevents the recycling of this receptor from the Golgi to the membrane. Nef has highly conserved protein–protein interaction domains essential for these functions (57). In addition, the Nef signature motifs used to downregulate MHC-1 are also used to downregulate CXCR4 and CCR5, which decreases the chances of HIV superinfection (18,171).
HIV reservoirs
HIV-infected individuals who are compliant to antiretroviral therapy (ART) show an apparent clearance of the virus in the peripheral blood shortly after initiating therapy. Nevertheless, viral particles can be detected after interruption of antiretrovirals (30,64,183). Moreover, there is genetic HIV evolution over time in patients with undetectable viremia. In addition, Buzon et al. used deep sequencing and statistics to demonstrate that in the presence of suppressive ART, the integrated HIV (i.e., archival, proviral HIV) and extrachromosomal HIV (episomal, surrogate for recent infection) belong to different viral populations (14), confirming continuous low-level replication of HIV.
Where does the virus hide? At the cellular level, resting T-lymphocytes (memory cells) or long-lived myeloid cells (macrophages and DC) remain transcriptionally silent for long periods of time while having integrated copies of the HIV genome, especially in the presence of potent antiretrovirals (22,23,41,71). The activation of these cells resumes the production of infectious particles, and hence the story repeats itself by [infection of new cells] ↔[reseeding of the reservoir] ↔[return to resting state] and perpetuation of the persistence of HIV. At the organ/system level, anatomic compartments that may serve as reservoirs of HIV include the central nervous system (28,96), the genitourinary tract (32,125), and the gut-associated lymphoid tissue (24,133). In the respiratory system, the lung can embrace high levels of HIV replication. Early studies showed that bronchoalveolar cells of AIDS patients may host at least 7.6-fold more HIV proviral DNA than cells in the peripheral blood (25). More than a decade later, Brenchley's group found that the frequency of HIV-infected BALc was comparable to that in blood cells (12). Studies with rhesus macaques infected with simian immunodeficiency virus (the simian homolog of HIV) showed that the lungs and the intestines harbored the highest levels of viremia, second to lymph nodes, even during the ART-induced suppression of blood viral loads (78).
In the alveoli, lymphocytes are more susceptible to HIV infection than macrophages. HIV infects 1 in 100 CD4+ alveolar lymphocytes (82) and 1 in 1,000 alveolar macrophages (AM) (26). The alveolar space harbors small and large subpopulations of macrophages, which differ not only in morphology but also in cell surface markers (54,178). HIV preferentially infects the small macrophages, which display features of highly active inflammatory cells (81). Although at lower levels than blood monocytes, alveolar macrophages express CD4, CCR3, CCR5, and CXCR4, which are receptors required for HIV infection and can be infected by several HIV isolates but not by CXCR4-utilizing viruses (175). HIV also infects CD8 T-cells and fibroblasts (105,143).
May the lungs act as anatomical reservoirs for HIV? Studies in the 1990s showed significantly less HIV sequence variability in AM leading to complete phylogenetic separation of the HIV lineages in the lung and the blood (121). Further genetic studies of the third variable domain (V3) of the HIV envelope showed tissue-specific quasispecies in the lungs, brain, and testis of AIDS patients (168). In the same studies, HIV isolates from the lung clustered with macrophage-tropic/nonsyncytium-inducing (NSI) variants in the blood, while isolates from the lymph nodes clustered with lympho-tropic/syncytium-inducing (SI) variants in the blood. Because both NSI and SI variants were detected in different tissues, the existence of tissue reservoirs apart from the brain was discounted, and the presence of HIV in nonlymphoid tissues was seen as a late-stage event secondary to lymphocyte infiltration (168). By using functional env clones from primary isolates, Singh's group (151) examined the HIV co-receptor utilization patterns in the lung and showed that the lung isolates used CCR5 as exclusive co-receptor, while the blood isolates may use either CCR5 or CXCR4. Furthermore, lung and blood isolates were undistinguishable regarding the use of secondary co-receptors, including CCR2b, CCR3, CCR8, and CX3CR1 and orphan receptors GPR1, GPR15, and STRL33, suggesting that co-receptor utilization does not determine HIV compartmentalization in the lung, despite the biological differences between HIV quasispecies in the blood and the lung.
Later studies that compared HIV env sequences spanning the second constant and the fifth variable region (C2–V5) from matched blood and lung samples (either lung sputa or BALc) found lung-specific evolution in up to 56% of HIV-infected individuals (74). In this study, HIV compartmentalization in the lung was supported by significant statistical comparisons but not by phylogenetic tree inspections, which showed only modest compartmentalization. This study also found almost identical sequences in BALc samples of a single patient with the greatest compartmentalization; the authors attributed these observations to localized clonal expansions of the viral quasispecies (74).
The lung is a macrophage-rich tissue, and hence myeloid cells represent strong candidates as cellular sources of HIV in the lungs. The idea that AM differentiate from precursor peripheral blood monocytes has been used as a basis in several HIV molecular evolution studies. Itescu et al. performed phylogenetic analyses of HIV envelope V3 loop sequences from circulating monocytes and lung macrophages of four HIV-infected individuals with diverse pulmonary diseases. They found highly homogeneous sequences in the BALc compared to the peripheral blood, suggesting a lack of unrestricted bidirectional trafficking of HIV quasispecies in the lung and therefore independent evolution (80). In the blood, HIV quasispecies in dendritic cells have different phylogenetic clustering patterns than those in their precursor blood monocytes (170). In both cases, the authors concluded that viral forms in either the lung macrophages or blood dendritic cells evolved independently from their blood monocytic putative ancestries. One caveat that might account for these observations, at least in part, is molecular sampling. At the time of sample collection, precursor monocytes in the blood and their differentiated versions of lung AM or blood dendritic cells may not correspond to the same generation of cells or, even more likely, the differentiated versions of the monocytes collected previously may have ended up in anatomical sites other than the lung or the blood. Also, the HIV infection of macrophages may have occurred postdifferentiation, especially in the presence of ART (3). Importantly, cell-to-cell contact between lymphocytes, dendritic cells, and macrophages that cohabit in the lung may certainly facilitate the spread of HIV quasispecies within the intrapulmonary neighborhood.
The existence of HIV reservoirs demonstrates that ART does not eliminate from the host. Although evidence points to lung-specific viral evolution, the lung is not as anatomically enclosed as the brain, and hence some viral leakiness is most likely to occur due to the active blood flow through the cardiopulmonary system. Although this shakens the notion of the lung as a HIV reservoir, cryptic replication of HIV in the lung might contribute to immune disturbances leading to pulmonary complications, and hence, deserves close attention.
Evidence of HIV as a Silent Pathogen in the Lung
HIV is present in the alveolar compartment, but how much of it is detected and resented by the lung? Alveolar macrophages from HIV-infected individuals are classically activated and are fully competent to respond to Streptococcus pneumoniae (61,63), an etiologic agent for recurrent pneumonias in the HIV group. HIV-challenged AM but not blood monocytes or peritoneal macrophages also retain normal antifungal activity against Cryptococcus neoformans, as per findings by Cameron et al. (16). HIV itself may not impair the phagocytic activity of AM unless combined with cigarette smoking (48). AM from smokers exhibit a decreased internalization of C. neoformans, regardless of HIV disease stage (136), suggesting that smoking, which is a prevailing problem in the HIV-infected population (75), certainly complicates the immunologic landscape in the lungs.
While these studies suggest that AM are fairly resilient to HIV in the lungs, contrasting evidence suggest that HIV alters the pulmonary cell biology. Compared to HIV-uninfected counterparts, HIV-infected individuals have lower secretion of interferon-gamma (IFN-γ) and tumor necrosis factor-alpha (TNF-α) in the lung, significantly increased RANTES and lysozyme in the BALf (62), and marked cellular activation and accumulation of inflammatory mediators in the alveolar space, including increased HIV-specific CD8+ T-cells.
Transgenic expression of HIV in a rat model showed decreased expression of the beta subunit of the GM-CSF receptor in AM and impaired bacterial phagocytosis in vitro potentially mediated by pulmonary zinc deficiency (85). HIV also impairs the innate antifungal activity and toll-like receptor-4 (TLR-4) signaling in alveolar macrophages and significantly decreases the microbicidal effector cell function against Pneumocystic carinii (79,95,157), probably mediated by a significant reduction of mannose receptor-mediated binding (94). Nicol's group found decreased TLR-1 and TLR-4 surface expression in U1 monocyte cells in vitro and in AM ex vivo, accompanied by decreased production of TNF-α, supporting the notion that HIV predisposes the host to co-infections (124).
The presence of HIV in the lungs may also impact bystander pulmonary residence cells such as endothelial cells. Pivotal studies by Kanmogne et al. showed that endothelial cells in the pulmonary microvasculature do not express CD4, CXCR4, or CCR5 receptors on their surface, although the presence of CXCR4 and CCR5 mRNA, but not CD4, was demonstrated in human microvascular endothelial cells (87). Despite being resistant to HIV infection, EC remain susceptible to apoptosis when exposed to HIV proteins (65,87,128,172). Later, it was demonstrated that the lung endothelial cells strictly regulate the CXCR4 surface expression, which can be significantly induced by erythromycin in human lung microvascular endothelial cells in vitro and lung capillary endothelial cells in vivo (158). While EC may not represent a cellular source of HIV in the lung, they certainly remain susceptible to the cytopathic effects of HIV.
The impact of HIV on the pulmonary cell biology can also be exemplified by the interactions with co-receptors. While CCR5 may have protective roles in lung inflammation (66,93,127). CXCR4 and its natural ligand CXCL12 are linked to malignant outcomes such as progression and metastasis of osteosarcomas (104,182), breast cancer (118), colon cancer (119), prostate cancer (1), ovarian cancer (6), and non-small cell lung cancer (NSCLC) (21,36,179). Whether HIV interactions with the CXCR4 receptor in lung cells mediate similar outcomes warrants further studies.
In summary, the mere presence of HIV in the lung translates to decreased antimicrobial cytokines, increased inflammatory and cell activation markers, and impaired responsiveness of bronchoalveolar T-lymphocytes and macrophages, all of which may ease the colonization and persistence of potential pathogens. Additionally, interaction of HIV with CXCR4 receptors may invoke noxious outcomes beyond cell infection (e.g., malignant phenotypes). Together, these take us to the arena of HIV-associated pulmonary complications.
HIV-associated challenges in the lung
The lung is a frequent target organ for disorders associated with HIV infection, including chronic obstructive pulmonary disease (COPD), lung cancer, pulmonary arterial hypertension (PAH), fibrosis, and infections, as indicated by echocardiographic abnormalities, which are usually accompanied by systemic and/or pulmonary inflammation (34,117). The following section will focus on COPD, pulmonary arterial hypertension, and lung cancer as three main pulmonary complications associated with HIV infection.
COPD
COPD is characterized by limited expiratory airflow, and represents the fourth cause of death worldwide (176). With a complex pathophysiology, COPD is overrepresented in HIV-infected patients compared to uninfected counterparts, and represents a significant risk factor for hospitalization in this population (2,46,108). A study that followed a HIV-infected cohort for 4.5 years in Rigshospitalet, Denmark, reported that the HIV group presented decreased forced expiratory volume in one second, alveolar volume, and carbon monoxide diffusion capacity over time, with modest signs of COPD at the time of the report (97). This observation was predicted by smoking and not associated with ART in separate studies in Sassari, Italy, and Ontario, Canada (35,108). Another study suggested that HIV-COPD co-exists with increased pulmonary artery systolic pressures (PASP) or tricuspid regurgitant jet velocity (TRV), in association with inflammatory markers (sputum interleukin-8, peripheral interleukin-8, peripheral IFN-γ levels) and peripheral T-cell activation (117). Conversely, studies using a considerable large data set from the Multicenter AIDS Cohort Study and the Women's Interagency HIV Study reported that noninfectious diseases were more frequent in the pre-ART era, but authors did not find a significant risk for COPD (odds ratio [OR]=1.61) or lung cancer (OR=2.65) post-ART (58). This study relied on self-reported pulmonary diagnoses, which were not confirmed clinically, and hence findings from this study might have been masked by the presence of unrecognized COPD.
COPD patients display abnormal accumulation of CD8+T-cells in the lungs, which triggers the release of inflammatory cytokines and growth factors from macrophage and neutrophils (7,69). Lassitier et al. showed that transgene expression of HIV depleted glutathione (the most abundant free radical scavenger in the cells) and increased oxidative stress in the alveolar compartment, linked to alterations in the expression of tight junction proteins and impaired barrier functions of the epithelial cells in the alveoli (99). Further studies by Fan et al. pointed to inhibition of the nuclear factor (erythroid-derived 2)-like 2/antioxidant response element (Nrf2/ARE) pathway as mechanistic insights into HIV-mediated barrier functions and oxidative status (51). HIV-associated COPD also features low levels of glutathione in the lungs and in the peripheral blood (33). Together, this suggests that the presence of infectious pathogens such as HIV, coupled with abnormal inflammatory responses and oxidative stress, all contribute mechanistically to HIV-COPD.
Emphysema is a COPD characterized by apoptosis of epithelial and alveolar cells, with various degrees of inflammation. While cigarette smoking is a major cause of COPD/emphysema (38), HIV is another risk factor for COPD, independent of smoking. Patients with HIV, including nonsmokers, are susceptible to accelerated forms of emphysema that develops significantly earlier than smoking-associated emphysema (45). Alveolar macrophages in HIV+ smokers display upregulated matrix metalloproteinases, which may contribute to the development of emphysema at a younger age and with higher incidence than HIV +nonsmokers (66). Histologically, HIV is mostly found in the emphysematous regions of the lung, while very rare HIV+ cells are present in normal lung areas (180), suggesting a direct role of HIV and/or HIV proteins in emphysema. Clauss's group proposed the upregulation of the inflammatory cytokine endothelial monocyte activating polypeptide II (EMAP II) as a mechanistic explanation for the lung endothelial cell apoptosis (27) and that such upregulation is induced by gp120 signaling through the CXCR4 receptor and activation of p38 MAPK (65).
Relevant to chronic bronchitis, CXCR4 and HIV-X4 viruses are implicated in the overproduction of mucus and mucous cell metaplasia in human bronchial epithelial cells in vitro, via the CXCR4/α7-nicotinic acetylcholine receptor/ γ-aminobutyric acid (GABA)-A receptor axis (70). These results further support a potential role of HIV in the higher incidence of COPD.
Pulmonary arterial hypertension
Pulmonary arterial hypertension (PAH) is a rare disease in the general population, affecting one to two people per million individuals, but it occurs significantly more frequently in the HIV-infected population, regardless of gender, age, sociodemographic characteristics, duration of HIV diagnosis, or interventions with ART.
PAH is characterized by elevation of circulating inflammatory cytokines, atypical pulmonary vascular remodeling featured quasi-malignant phenotype of pulmonary endothelial cells (i.e., “cancer paradigm” of PAH) (101,135), and highly glycolytic pulmonary artery vascular cells (60,161). In addition, a signature of PAH is the presence of cells that obliterate the lumina of pulmonary arteries (plexiform lesions) (162). Therefore, the mean pulmonary artery pressures increase (mPAP >25 mmHg), ending fatally due to right heart failure. PAH can be screened by echocardiography, which measures PASP and diagnosed by right heart catheterization (RHC), which measures mPAP; final diagnosis is made based on mPAP >25 mmHg.
The prevalence of PAH in the HIV-infected population is usually reported to be 1 in 200 (0.5%) individuals. Nonetheless, the awareness of HIV-PAH has increased in medical communities worldwide, as evidenced by the coordination of taskforces aimed to screen patients who are asymptomatic for PAH. PAH has been recently reported to affect 0.2–12.7% of HIV-infected individuals in several countries (Table 1), based on either echocardiographic PASP or RHC. Noteworthy, Vazquez et al. found a single HIV+ patient in a cohort of 445 individuals with PAH (0.2%) and posed the question of whether all patients with PAH should undergo HIV testing (169). The opposite is also valid: should all patients with HIV undergo screening for PAH?
A study comparing pressures measured by Doppler-based echocardiography versus right heart catheterization showed that 19.7% of the Doppler-based measurements were inaccurate, missing the PAH phenotype in one out of three patients (142). Despite this, the reality is that many patients may just decline the RHC procedure, are ineligible, or it may just not be available, especially in low-resource settings. Hence, the best scenario in many instances is to retrieve echocardiography data and use PASP 30–35 mmHg as a cutoff for echocardiographic abnormalities associated with PAH, with the caveat that some of these data may still be underestimated but are at least accounted for.
The increased prevalence of HIV-PAH (whether accurate or underestimated) has been documented by several studies worldwide, but have we refreshed the hints for mechanisms and targets for therapy? Do we really know what in HIV increases the chances of PAH? There is no definitive proof that HIV causes PAH and no evidence that HIV infects lung endothelial cells (62). However, viral proteins and their interactions with molecular partners in the infected cells may damage endothelial cells, induce inflammation, and deregulate apoptosis and proliferation of vascular endothelial cells in the lung, resulting in pulmonary vascular remodeling featured in PAH patients (65,88,106,113,172). Previous studies have shown that HIV Nef protein co-localized with EC in PAH-like plexiform lesions in rhesus macaques infected with the SHIVnefSF33 chimera (expressing human Nef ) (110,111). Subcellular analyses showed increased cellular content of giantin and p115 (Golgi tethers) in cells in plexiform lesions, particularly in Nef-positive cells (141), suggesting a Nef-Golgi dysfunction duet in PAH. After translating the animal studies to humans, Nef signature sequences were found to be associated with the PAH phenotype (4). Separate studies in two macaque models (rhesus macaques infected with SIVΔB670 and cynomolgous monkeys infected with chimeric SHIV89.6P-env) also showed intimal and medial hyperplasia along with elevated pulmonary pressures resembling human PAH (56). Together, these studies suggest that HIV proteins play key roles in the pathogenesis of HIV-PAH.
The combination of HIV—and/or its proteins—and recreational drugs such as cocaine exacerbates pulmonary arteriopathies. Dhillon et al. showed that HIV Tat or cocaine disrupted tight junction proteins, increased the expression of platelet-derived growth factor, and increased the proliferation of pulmonary smooth muscle cells; additive effects where observed with combined Tat and cocaine (44). HIV Tat also increased the basal inflammatory status and oxidative stress in a murine model (31). Additionally, macaques exposed to the simian immunodeficiency virus (simian homologue of HIV) and morphine also showed significant pulmonary vascular remodeling and oxidative-stress mediated apoptosis of endothelial cells followed by proliferation of apoptosis-resistant cells (153). In the framework of pulmonary artery vascular smooth muscle cells, Goncharov et al. recently showed that the balance tilts toward increased proliferation and decreased apoptosis when the mammalian target of rapamycin complex-2 (mTORC2) downregulates the energy sensor AMP activated protein kinase (AMPK) (60).
In light of the absolute presence of inflammation in PAH, Tcherakian et al. proposes that a lack of CD4 T-cell restoration in the gut is followed by translocation of lipopolisaccaharides, which induces the overproduction of interleukin yielding to chronic inflammation (159).
PAH remains incurable. Current therapeutic approaches aim to prevent further pulmonary damage by using parenteral, inhaled, and oral combinations of prostaglandins (e.g., prostanoids), endothelin receptor antagonists, diuretics, calcium channel blockers, and phosphodiesterase-5 inhibitors (19,112,134). Hopes of reversing established PAH via regenerative approaches, including the use of endothelial progenitor cells and mesenchymal stem cells, await extensive clinical testing (155). Riociguat is a soluble guanylate cyclase stimulator that belongs to a new class of vasodilators currently in clinical development, which reversed right heart hypertrophy and ventricular remodeling associated with PAH in rats and mice (5). Moreover, telmisartan improved pulmonary artery EC dysfunction and remodeling in a monocrotaline rat model of PAH through the peroxisome proliferator-activated receptor γ (PPAR-g)-dependent P13K/Akt/eNOS pathway (103).
The unquestioned success of ART in controlling HIV and the impact of ART on PAH remains a dichotomy because some studies suggest that ART does not protect but rather accelerates the development of PAH (129,138) on top of drug–drug interactions (discussed below).
HIV-associated malignancies
HIV-associated malignancies that define AIDS (e.g., Kaposi's sarcoma, non-Hodgkin lymphoma, and cervical cancer) have decreased in the post-ART era. Nonetheless, the incidence of non-AIDS–defining cancers (NADC) have tripled. In a retrospective study at Casey House, Toronto, Canada, up to 52% of deaths in the post-ART era were ascribed to NADC, including liver, gastric, colorectal, and lung malignancies, which occurred in patients with fairly well-controlled HIV disease (154). In Cologne/Bonn, NADC accounted for the death of about one in six HIV-infected people (47). Post-ART, people living with HIV in California have a high cancer risk, as shown by significantly higher rate ratio (RR) for anal (RR=56), liver and melanoma (RR=2), lung (RR=1.2), and prostate and colorectal (RR=1) cancers (150). In patients across Europe, Israel, and Argentina (EuroSIDA), lung cancer (9%) was the second most common NADC after anal cancer (19% vs. 9% respectively) (137).
Globally, lung cancer is the most common cancer (59). In turn, lung cancer is more frequent among HIV-infected individuals in the era of suppressive ART (72). A study that encompassed 15 years pre- and post-ART documented increases in NADC including prostate cancer (11% increase), anal cancer (13% increase), liver (20% increase), and lung cancer (46% increase) (148). The most common type of lung cancer in the HIV-infected population is the adenocarcinoma (149), although NSCLC was found in 88% of cases with HIV-associated lung cancer (72). Compared to the HIV-uninfected population, patients with HIV present with a younger age (mean 50 years) at the time of diagnosis with lung cancer (40). In addition, most of the affected patients are smokers and unfortunately present with symptoms of advanced cancer (144). HIV itself is an independent risk factor for lung cancer, regardless of smoking, COPD, and bacterial pneumonia (149).
One of the mechanisms proposed for HIV-associated lung cancer is immunosuppression (68,137,150). Meta-analyses from Shiels's group reported an increased incidence of lung cancer among patients with AIDS-related immunosuppression (146). Separate studies found a 2.2 relative risk of lung cancer in HIV-infected immunosuppressed patients (with CD4 counts <200 cells/mL) compared to uninfected individuals (150). Contrasting data from several groups show that lung cancer in HIV-infected individuals is not associated with CD4 counts (20,91,147), despite the inverse relationship with HIV viral load (50). Results from the D:A:D Study group, which followed >40,000 HIV-infected individuals over 200 clinics in Europe, the United States, and Australia from 2004 to 2010 also identified lung cancer as the most common NADC, with the highest mortality rate, regardless of CD4 counts (177).
Additional mechanistic views into lung malignancies associated with HIV infection are provided by respiratory infections and genomic instability. The HIV-infected population is particularly prone to bacterial pneumonia, in addition to mycobacterial pneumocystis, and viral respiratory infections. Pulmonary infections, in turn, may increase the risk of lung malignancies in the HIV-infected population (49,91,145). In addition, genomic instability, reflected by microsatellite alterations, has been hypothesized to increase the risk of lung cancer in HIV. Microsatellite alterations, but not the loss of heterozygosity, were significantly increased (sixfold higher) in HIV-associated lung carcinomas (173). What produces this HIV-related genomic instability? A previously unrecognized interaction between HIV and endogenous retrotransposable elements has been uncovered with the finding that HIV infection results in accumulation of type 1 long-interspersed nuclear elements (L1s) DNA in primary CD4+ lymphocytes (84).
Inflammation is also an ingredient in the recipe for HIV-associated cancers. A study that evaluated activated inflammatory pathways (IL-6 and C-reactive protein) and coagulation pathways (D-dimer) in HIV-infected patients found that individuals with higher levels of IL-6 had a significantly higher risk for cancer (11).
Despite the potential underestimations due to calculations based on self-reported disorders, the use of screening tools for diagnosis, or even undocumented patient cohorts, the higher susceptibility of HIV+ patients to serious lung complications including COPD, PAH, and lung cancers is evident. Inflammation, oxidative stress, deregulated apoptosis and proliferation, and malignant phenotypes are common denominators in the quest for mechanistic hints. Many of the specifics regarding the direct and indirect role of the virus in these diseases remain undetermined.
Success and Challenges of Antiretrovirals
Combinations of antiretrovirals, introduced in 1996, reformed the standard of care for HIV patients and antiretroviral drug industry. The Food and Drug Administration in the United States has 33 drugs currently approved for HIV treatment (165). Current ART targets HIV entry, fusion, reverse transcriptase, integrase, and proteases. In the setting of HIV receptors, Maraviroc is the only CCR5 receptor blocker available since 2007. Plerixafor antagonizes the CXCR4 receptor and mobilizes hematopoietic stem cells (166). It has been available as a chemotherapeutic agent since 2008, but its feasibility for anti-HIV treatment was rejected after reports of serious thrombocytopenia, premature ventricular contractions, and paresthesias (76). Other compounds, including TG-0054 (for chemotherapeutic purposes) and AMD-070 (for anti-HIV uses), are being tested in clinical trials (37). While pre-exposure prophylaxis and/or heavy postexposure prophylaxis represent robust hopes for a cure nowadays (9), research interests are now trending toward a functional eradication of HIV. This strategy aims for a profound suppression—not eradication—of the virus, particularly in reservoirs, so that patients can discontinue antiretrovirals, which will require extensive knowledge about potentially unrecognized reservoirs and reservoir modulation as a solid platform.
ART significantly decreases the HIV viral load in the bronchoalveolar lavage fluid to undetectable levels in >80% patients; it also decreases IFN-γ, IL-6, and CD8+ lymphocytes while increasing CD4+ cells in the alveolar space, returning the BAL differential toward normal values (92,163,164). The reconstitution of the CD4+ cell pool appears to occur via proliferation of lung resident CD4 cells rather than recruited from the periphery (92), which might have implications in the reseeding of the HIV reservoirs in the lung. Together, ART significantly decreases the HIV viral load and cell activation in the lungs and decreases inflammatory cytokines. However, other studies report that HIV impairs TLR-mediated TNF-α production in the alveolar compartment, decreases in TLR-1 and TLR-4, leading to deficiencies in innate immune response in the presence of ART (124). In addition, measurements of glutathione and cysteine levels in BAL as surrogate markers of oxidative stress in the lung showed no significant differences between HIV-infected and uninfected subjects and that HIV-infected patients without ART had significantly decreased alveolar glutathione levels, suggesting increased oxidative stress in that population (33).
Given the significant reduction in mortality accomplished by ART, the HIV-infected population lives as long as the noninfected. A 15-year longitudinal study by Moore et al. (116) in Maryland reported that the expected longevity of HIV-infected patients is now 73 years. A separate study using a stochastic computer simulation model of HIV infection in the United Kingdom calculated that HIV-infected individuals have a life expectancy of 72 years nowadays in the ART era (120). Given that the global life expectancy at birth in 2011 was 70 years (range of 56 years in Africa and 76 years in America, Europe, and Western Pacific) (176), these studies confirm that antiretrovirals have rendered HIV as a chronic disease, especially in developed countries (39).
Nevertheless, the benefits of ART come with side effects, including insulin resistance (52), lypodystrophy (43), osteoporosis (140), subfertility (98), and atherogenic chances including EC dysfunction, neointimal hyperplasia, and mitochondrial damage (83). ART also has an impact at the pharmacodynamic level via interactions with antituberculosis agents, antimalarials, anticoagulants, chemotherapeutic agents, and pulmonary antihypertensive agents, especially when co-administered with newer antiretrovirals, including darunavir (protease inhibitor), raltegravir (integrase inhibitor), maraviroc (CCR5 antagonist), and etravirine (non-nucleoside reverse transcriptase inhibitor) (89,131,132,152). Given the number of comorbidities along with HIV disease, it is almost inevitable to avoid the co-administration of medications and the complexity that it involves in the developed world.
According to the World Health Organization, 9.7 million HIV+ people were receiving the life-saving ART in low- and middle-income countries at the end of 2012, 77% of whom live in the African region. This means that the current HIV ART coverage in Africa has tripled since 2008 (176). Now that the ART era is being extended geographically to Africa, noninfectious complications of HIV infection would be a foreseeable challenge, just like in the developed world. Here is a different one though: immune reconstitution inflammatory syndrome (IRIS). IRIS occurs in response to residual pathogens including, but not limited to, Cryptococcus, nontuberculous mycobacteria, pneumocystis, and tuberculosis (15). The sub-Saharan African population is facing high rates of mortality within months of initiating ART, with 73% of the cases due to IRIS (174), while other studies report high morbidity that is easy to manage therapeutically, but low mortality (15,100). In view of this, timing for therapeutic interventions and dose adjustments are being considered to resolve this dilemma. Current considerations involve starting antiretrovirals 2 weeks postinitiation of antimycobacterials in patients with CD4 counts >50 cells/μL (100,102).
Another aspect to contemplate is the fact that most of the antiretrovirals known have been developed and tested in subtype B HIV strains, which prevail mostly in America, Western/Central Europe, Southeast Asia, Northern Africa, the Middle East, and Australia (13). Non-B subtypes of HIV exhibit resistance to drugs effective on subtype B (73). For instance, a study on antiretroviral-naïve HIV-infected individuals with subtypes F, G, and K/F, A/F, and J/F recombinants in Romania found resistance mutations in the pol gene in up to 50% of cases (160). In Tanzania, 12% of antiretroviral naïve HIV+ pregnant women infected with subtype A and C presented primary drug resistance (167). After exposure to ART, up to 51% of cases had a positive drug resistance genotyping in KwaZulu-Natal, South Africa, where subtype C predominates (109). In Cote d'Ivoire, West Africa, up to 25% patients infected mostly with A/G recombinants acquired mutations to at least one drug in the antiretroviral cocktail (114). In Spain, up to 19% of the patients infected with non-subtype B recombinants displayed resistance mutations in the pol gene (181). On the other hand, a study in France found that patients infected with B and non-B subtypes of HIV have similar virologic (HIV RNA levels <50 copies/mL) and immunologic responses (CD4 counts) to antiretrovirals (17). Viruses of different subtypes may also be comparable in terms of antiretroviral drug susceptibility. For instance, studies using structural models of the HIV integrase enzyme showed that subtype B viruses and recombinant A/G viruses (prevalent recombinant form in West Africa) have comparable susceptibility to integrase inhibitors (123). Kenya seems to have a very promising drug resistance profile: drug resistance mutations were detected in <2% of antiretroviral-naïve individuals (90). Hence, the expanded availability of ART must be accompanied by initial HIV subtyping and profiling of primary and acquired drug resistance mutations as much as possible so that efficient antiretroviral therapies can be tailored accordingly.
Viewpoints and Potential Avenues for New Research
While ART has been a significant milestone in the battle against AIDS, we are still learning about HIV and facing challenges 30 years after its discovery. Figure 1 outlines key concepts discussed in this review. This review illustrated key concepts of HIV persistence and focused on how the lung resents HIV, echoing HIV-associated pulmonary complications such as COPD, pulmonary arterial hypertension, and lung malignancies.
A systematic epidemiological surveillance is strongly desirable to document HIV-associated pulmonary complications. Therefore, cross-talk between pulmonologists and HIV specialists is essential to document the true prevalence of these diseases, which otherwise would go unnoticed and untreated before the patient's quality of life has seriously deteriorated. Antiretroviral drug toxicity, resistance, and drug–drug interactions are issues affecting both the developed and the developing world, which certainly require extensive research and implementation of revised therapeutic strategies.
This article also presented empirical data that will hopefully generate new hypotheses for new research enterprises. In the first instance, the increased production of mucus and the risk for cancers implicated in the HIV-X4/CXCR4 axis evidence a profound impact of HIV in the pulmonary cell biology and invoke the notion that HIV-X4 viruses are worth investigation in the context of COPD and lung cancer. Second, further studies to dissect the role of HIV and HIV proteins in PAH, particularly at the subcellular level and the impact in cellular cross-talk (e.g., EC, macrophages, T-cells, SMC) are warranted to identify novel therapeutic targets. Third, it is necessary to develop antiretroviral compounds that are more suitable to non-B subtypes of HIV. Fourth, the identification of potentially unrecognized anatomical reservoirs for HIV, as well as knowledge about the modulation of cellular sources of viruses within reservoirs, are absolutely required to strategize for a functional eradication of HIV reservoirs. Functional eradication of HIV reservoirs in the lung would increase alveolar macrophage and bronchoalveolar lymphocyte responsiveness and decrease the likelihood of co-infections with potential pathogens. Finally, the role of chronic inflammation in HIV-associated pulmonary diseases is certainly a unifying, hypothesis-generating topic that will take us to the next level.
Footnotes
Author Disclosure Statement
No competing financial interests exist.