
Abstract
Hepatitis B and C (HBV and HCV) are hepatotropic viruses in humans with approximately 350 and 170 million chronic carriers respectively. Since both viruses have similar modes of transmission, many people are co-infected. Co-infection is common in intravenous drug users, HIV-positive individuals, and transplant recipients. Compared to mono-infected patients, co-infected patients exhibit exacerbated liver cirrhosis, hepatocellular carcinoma, and liver failure. Some of the pathogenic effects may be attributed in part to the structural core proteins of both viruses—ones that have displayed immunomodulatory properties. Yet, the effects of their combined interaction on the human immune system remain a mystery. We aimed to elucidate the combined effects of HBV and HCV core proteins on human dendritic cells' (DCs) ability to present antigens and stimulate antigen-specific T-cells. We observed that when DCs, differentiated from human peripheral blood monocytes, were co-incubated with both core proteins, IL-10 production was dramatically enhanced, IL-6, TNF-α, and IL-12 production was significantly reduced, and HLA-DR expression was downregulated. This instant functional and phenotypic modulation of DCs induced by a combination of HBV and HCV core proteins can allow them to behave like tolerizing DCs, inefficiently presenting antigens to CD4+ T-cells and even suppressing induction of the cellular immune response. These results reveal an important mechanism by which HBV and HCV synergistically induce immune tolerance early in infection that may be instrumental in establishing chronic, persistent infections.
Introduction
W
Both viruses can be transmitted from mother to child perinatally, through shared needles during intravenous drug use, and by improperly screened blood transfusions (39,40). HBV can also be transmitted through sexual contact (39). Given their similar modes of transmission, co-infection of HBV and HCV is common, although its prevalence is unknown (11). Between 2% and 10% of anti-HCV-positive patients also test positive for HBsAg, while 5–20% of patients with chronic HBV infection test positive for anti-HCV-antibodies (11). However, an inherent difference exists in the natural history of infection with either virus. HBV results in a self-resolving acute infection in adults, whereas infection of adults with HCV commonly leads to chronic, persistent infection (33). While acute infection with both viruses leads to unmodified infections relative to single infection, superinfection of chronic HBV patients with HCV leads to chronic HBV/HCV infection and exacerbated liver diseases (11). Regardless, co-infection significantly increases the risk of developing end-stage liver diseases, liver failure, and hepatocellular carcinoma (HCC) (5,8,42).
There is much debate behind interviral interactions and their role in increasing the risk of HCC in co-infected patients. Studies have shown the inhibition of HBV by HCV in co-infected individuals (27,32,36) or vice versa (28,35); others, however, have shown no interaction (4).
Downregulation of cellular immune responses plays an important role in the establishment and severity of co-infection. Several investigations have shown that various proteins of both viruses have roles in downregulating innate and adaptive immune responses (9,15,17,28,35). Antigens of both HBV and HCV have been shown to inhibit TLR-mediated signaling (24,41).
The core proteins of HBV and HCV have demonstrated distinct roles in hindering the development of adaptive cellular immune responses. While development of T-cell responses against HBV core has been associated with viral clearance (7), HCV core has been shown to modulate both innate and adaptive immune responses (14). Transfection of human CD4+ T-cells with HCV core protein increased expression of regulatory T-cell markers CTLA-4 and Foxp3, reduced production of immune-stimulatory cytokines IL-2 and IFN-γ, and increased production of IL-10, an anti-inflammatory cytokine (1). The regulatory function of these T-cells was confirmed, since they inhibited the proliferation of bystander CD4+/CD8+ T-cells (1). Moreover, they assumed an anergic phenotype, becoming unresponsive to T-cell receptor (TCR) stimulation (1). Additionally, HCV core protein can bind to complement component C1q, downregulating IL-12 production from macrophages and dendritic cells (DCs) (26), and has also been shown to downregulate NF-kB activation when modulating DCs (24).
In co-infection, core proteins of both viruses may interact together to affect immune responses. However, the combined effects of the two viral core proteins on the innate immune system's ability to activate cellular immunity have not yet been studied. We therefore examined the effect of co-culturing HBV and HCV core proteins on human DCs in vitro to elucidate the consequences of co-infection on the innate immune system. Incubation of DCs with core proteins of HBV and HCV together significantly modulated cytokine (TNF-α, IL-6, IL-10, and IL-12) production and MHC class II HLA-DR expression when compared to DCs incubated with core proteins of HBV and HCV individually. The functional and phenotypic modulation of DCs may affect their ability to stimulate antigen-specific T-cells, reducing the recognition of virus-infected host hepatocytes and viral clearance.
Methodology
Preparation of human peripheral blood monocyte derived DCs
Peripheral blood samples were obtained from HBV and HCV-negative donors (30–60 years of age of both sexes) from a phlebotomy clinic after written informed consent was obtained. Use of human blood samples and the written consent form were approved by the Health Research Ethics Board at the University of Alberta, Canada. DCs were generated from human peripheral blood mononuclear cells (PBMCs) as described previously (1,24,26). Briefly, PBMCs were isolated from blood by Ficoll-Hypaque (Amersham Biosciences) density gradient centrifugation. The intermediate buffy layer containing PBMCs was collected, and 5 × 106 cells/mL were cultured for 2 h in 6-well plates in RPMI 1640 medium (Invitrogen Life Technologies), supplemented with L-glutamine, 1% human AB serum (Sigma-Aldrich), 1% sodium pyruvate (Invitrogen Life Technologies), and 500 U/mL penicillin/streptomycin (Invitrogen Life Technologies). The adherent cells were treated with 50 ng/mL granulocyte macrophage colony-stimulating factor (GM-CSF) and 10 ng/mL of IL-4 (Peprotech) in RPMI media and cultured for 6 days. On day 6, >95% of the obtained cells were positive for both CD-11c and HLA-DR, suggesting differentiation of myeloid DCs.
Culturing DCs with viral core proteins
The immature, differentiated DCs were incubated with either HBV core (HBVc; 1 μg/mL); HCV core (HCVc; 1 μg/mL); HBVc (0.5 μg/mL)+HCVc (0.5 μg/mL); or recombinant human superoxide dismutase protein (rh-SOD; 1 μg/mL) for 24 h. Culture supernatant and cells were then collected for cytokine and flow cytometry analyses. The experiments were repeated with five individual donors. The recombinant HBV core was purchased from Prospec Bio. The HCV core protein and SOD control protein were gifts from Novartis. All of the control and recombinant proteins were tested for the presence of endotoxin using the Limulus amebocyte lysate (LAL) test kit (Genscript), and were found to contain <0.4 EU endotoxin/10 μg.
Flow cytometry
Cell suspensions containing 5×105 DCs were stained for 30 min on ice using our previously reported procedures with the following fluorescently conjugated monoclonal antibodies: anti-HLA-DR-QR (Sigma-Aldrich), anti-HLA-ABC-FITC, anti-CD11c-PE, anti-CD95-FITC, anti-CD86-APC, anti-CD40-FITC, and appropriate isotype control mouse IgGs (eBioSciences) (1,26). Samples were read on FACSCANTO and analyzed using FACSDIVA software (BD Biosciences). The cells were gated on the basis of side and forward scatter and then selected for CD-11c positive cells. More than 95% of the cells were positive for CD-11c, confirming the DC phenotype of the preparation (data not shown). Markers and gates were set to exclude >98% of corresponding isotype control antibody-stained cells.
Cytokine enzyme-linked immunosorbent assay
Cytokines (IL-6, IL-10, IL-12, and TNF-α) secreted in the culture supernatant of DCs treated with recombinant HBV and HCV core proteins were quantified using Ready-Set-Go enzyme-linked immunosorbent assay (ELISA) kits (eBioscience). A dilution of 1:2 to 1:20 was used for the samples, with the standards ranging from 5 to 2,000 pg/mL. The ELISA plates were finally read, and the concentrations were calculated with an automated ELISA plate reader (Fluostar Optima, BMG Labtech).
Statistical analyses
Error bars represent mean±standard deviations (SD) of data from three to five different experiments with individual donors. Statistical analysis was performed by one-way analysis of variance (ANOVA) and t-tests using Graphpad Prism (Graphpad Software Inc.).
Results and Discussion
We quantified the cytokine milieu produced by human DCs incubated with HBV and HCV core proteins as a model of how co-infection might modulate the immune response to these viruses. HBVc, HCVc, or SOD (total protein concentration of 1 μg/mL) were incubated for 24 h as an initial measure of the immediate effects of co-incubation on innate immunity.
Incubation with HBVc, HCVc, and HBVc+HCVc proteins significantly inhibited the production of TNF-α, a pro-inflammatory cytokine, when compared to the untreated group and SOD protein-treated group (p<0.05; Fig. 1A). However, HCVc, which completely suppressed TNF-α production (below detection levels), appeared more active than HBVc (179 pg/mL). Interestingly, TNF-α levels remained undetectable when HBVc was added to HCVc, suggesting that HCVc may exert a more dominant effect than HBVc in impairing the inflammatory activities and maturation of DCs in co-infection.
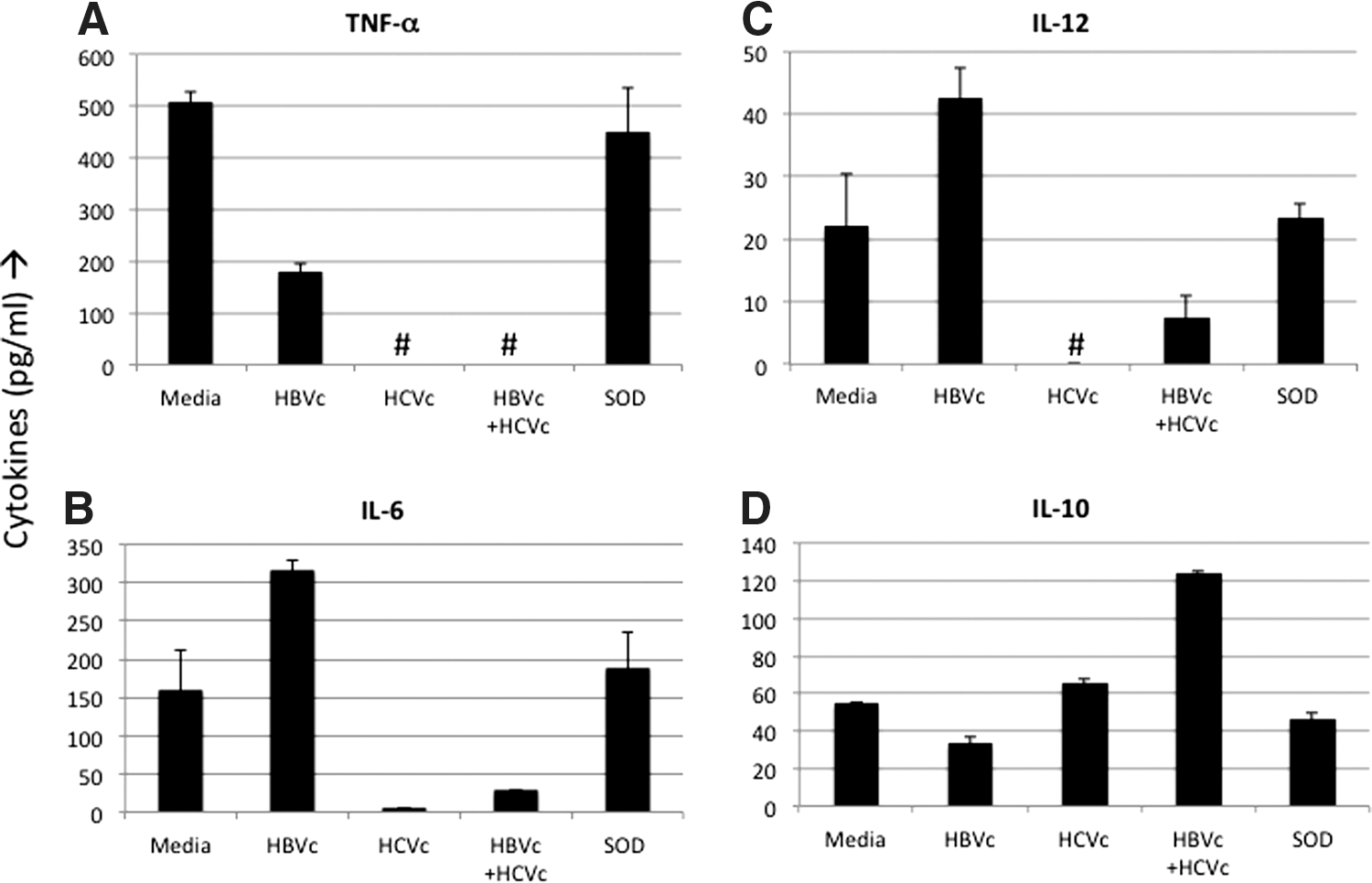
Reciprocal modulation of pro-inflammatory and inhibitory cytokines by combined core proteins of hepatits C virus (HCV) and hepatitis B virus (HBV).
IL-6 demonstrated a different pattern (Fig. 1B). Incubation with HBVc increased levels of IL-6 (317.5 pg/mL) when compared to the untreated group and SOD protein-treated group, suggesting that HBVc is pro-inflammatory. In contrast, incubation of DCs with HCVc almost completely stopped the production of IL-6 when compared to the untreated group and SOD protein-treated group. The reduction in IL-6 levels was statistically significant when compared to both untreated and SOD groups. These results suggest that HCVc has immune-regulatory properties. IL-6 levels in co-incubated DCs were also reduced and were significantly lower than DCs treated with HBVc alone or SOD protein alone (p<0.05). IL-6 production was, however, not significantly lower when compared to HCVc-treated DCs. Ultimately, co-incubation of DCs with HCVc exerted a negative effect on IL-6 production, again suggesting that HCVc dominates HBVc and that various biological activities in immune regulation and inflammation are impaired by co-infection.
Production of IL-12 appears to increase when DCs are incubated with HBVc (42.5 pg/mL) when compared to untreated DCs (22 pg/mL) and DCs treated with SOD protein (23.5 pg/mL; Fig. 1C). However, the increase is statistically insignificant. Incubation of DCs with HCVc completely abrogated the production of IL-12 (below detectable levels) compared to untreated and SOD groups as was seen for TNF-α and IL-6 production. These results further support the notion that HCVc is immune regulatory. Interestingly, compared to the HBVc group, both HCVc and HBVc+HCVc groups had significantly lower IL-12 levels.
HBVc evidently increases inflammation by stimulating DCs to produce IL-6. However, HCVc inhibits inflammation by downregulating levels of TNF-α, IL-6, and IL-12. Combining HBVc with HCVc exerts a predominantly anti-inflammatory response by decreasing production of TNF-α, IL-6, and IL-12. Co-incubation with core proteins from both viruses may prevent DCs from activating the adaptive immune responses.
Notably, levels of IL-10, an immune-regulatory cytokine, were increased by incubation with HCVc (66 pg/mL) and HBVc+HCVc (124 pg/mL; Fig, 1D). In contrast, IL-10 production was decreased by incubation with HBVc (33.5 pg/mL) when compared to untreated and SOD protein-treated controls. These results were statistically significant when compared to untreated DCs and those treated with SOD protein (p<0.05). These findings further confirm HBVc's pro-inflammatory nature and the suppression of a DC-activated adaptive immune response by HCVc and HBVc+HCVc.
Cytokine analysis shows us that co-incubation of viral core proteins inhibits DCs' function through two mechanisms: by preventing them from activating the immune system and by causing them to suppress it actively.
DCs also affect the stimulation of adaptive cellular immune responses through the expression of various cell surface markers. We examined expression of HLA-ABC, CD11c, HLA-DR, CD86, CD40, and CD95 on DCs co-cultured with HBVc and/or HCVc proteins. The most significant difference was observed in HLA-DR expression (Fig. 2A). DCs incubated with HBVc, HCVc, and HBVc+HCVc appeared to express lower levels of HLA-DR when compared to untreated and SOD protein-incubated DCs. Statistically, however, only downregulation was significantly affected by co-incubation when compared to untreated SOD protein-treated DCs (p<0.05). Based on mean fluorescence intensity and percent positive cells, the expression of CD11c and HLA-ABC (Fig. 2B and C), and CD40, CD86, and CD95 (data not shown) were not affected in DCs incubated with HBVc, HCVc, or HBVc+HCVc when compared to untreated or SOD-treated controls.
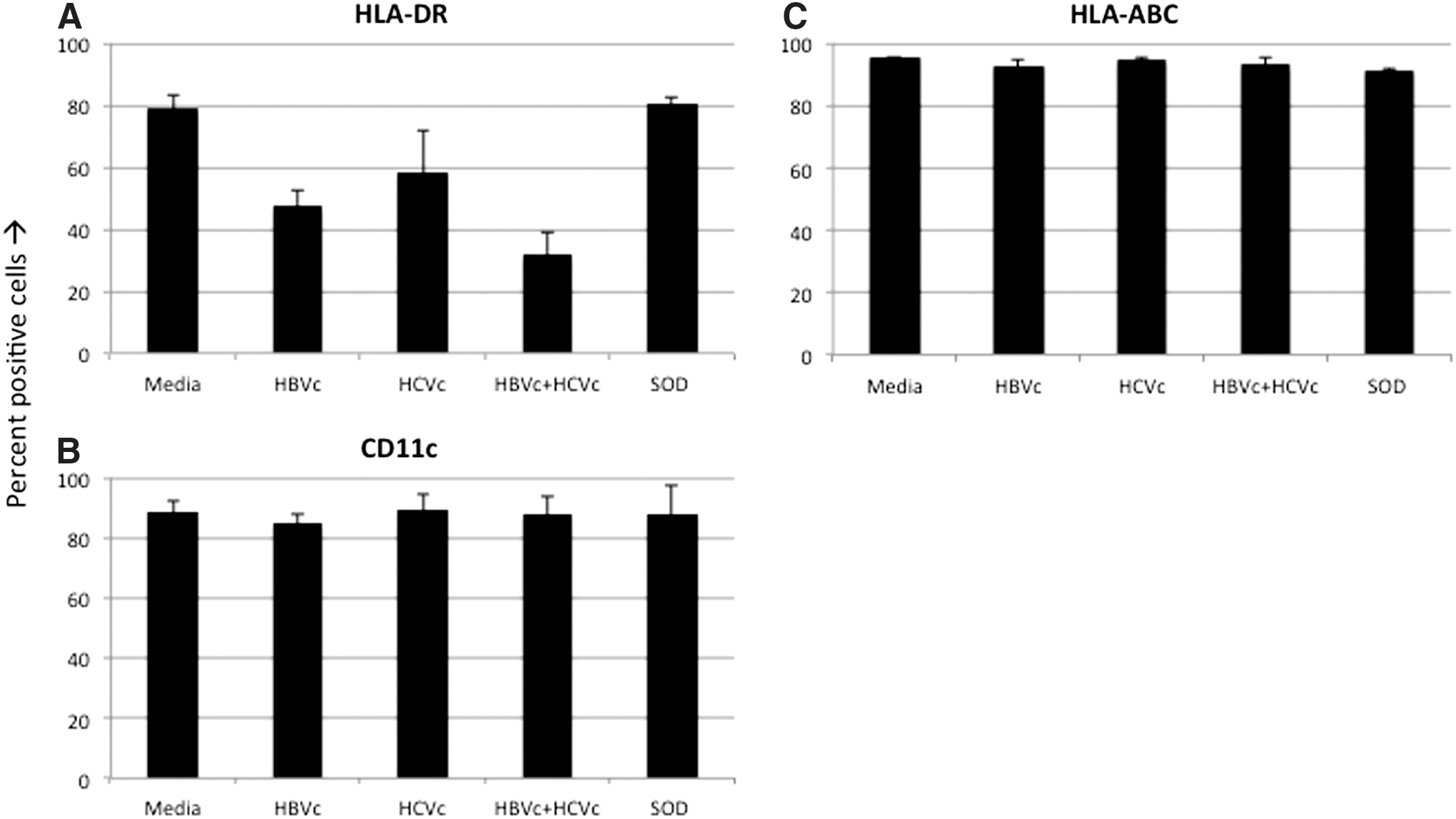
Combined core proteins of HCV and HBV modulate the antigen presentation capability of DCs through MHC class II expression:
Our results demonstrate an instant and significant effect of core proteins of HBV and HCV on DCs' ability to activate cellular immunity. Co-incubation with HBVc+HCVc modulates immune function in two ways: it decreases immune activation and increases immune suppression. We show that viral core proteins synergistically inhibit DC-induced immune activation by modulating the expression of both pro-inflammatory and inhibitory cytokines, and by inhibiting expression of DC surface molecules important for priming CD4+ T-cells. Specifically, IL-10 production was stimulated by co-incubation, while production of TNF-α, IL-6, IL-12, and HLA-DR were significantly inhibited by co-incubation. Interestingly, while TNF-α levels were decreased by incubation with HBVc, TNF-α levels were undetectable upon mono- and co-incubation with HCVc.
Co-incubation of DCs with core proteins also upregulated the production of IL-10, an immune-suppressive cytokine (19,31). Initially described as “cytokine synthesis inhibitory factor” (CSIF) when identified by Mossman et al. (18), IL-10 has been shown to inhibit expression of pro-inflammatory cytokines such as TNF-α, IL-6, and IL-12 (31). Chung et al. show that macrophages and DCs stimulated with Borrelia burgdorferi produce high levels of IL-10 and low levels of TNF-α, IL-6, and IL-12 in vitro (12). However, levels of pro-inflammatory cytokines were significantly higher in macrophages and DCs from IL-10−/− mice (12). Thus, core protein-mediated immune suppression may stem solely from the stimulation of IL-10, which subsequently suppresses expression of pro-inflammatory cytokines such as IL-12, IL-6, and TNF-α. Excess IL-10 has also been shown to induce anergy in antigen-specific CD4+ T-cells (23). As these antigen-specific T-cells become anergic, they likely suppress further T-cell proliferation (37), blunting the immune response against both viruses. In fact, previous studies have shown that viral core proteins also act similarly on CD4+ T-cells, causing them to express inhibitory molecules and become anergic (17). Moreover, secretion of IL-10 from DCs has been shown to induce regulatory T-cell type 1 (Tr1) cells (22), a subset of T-cells capable of actively suppressing cellular immune responses. Gregori et al. identified a subset of IL-10 producing DCs, which were termed “DC-10s” (22). DC-10s express high levels of CD40, CD80, and CD86 (22). Since our flow cytometry analyses show little change in surface expression of CD40, CD80, and CD86 (data not shown), we cannot conclude that co-incubation of naïve DCs with HBVc and HCVc causes them to differentiate into DC-10s. However, co-incubated DCs, such as DC-10s, produce high levels of IL-10, suggesting a role in inducing Tr1 cells. These Tr1 cells may further blunt the immune response against core proteins, limiting viral clearance.
Restricted HLA-DR (MHC class II) expression upon co-incubation may also be a consequence of overexpression of IL-10. Chung et al. (12) showed that macrophages, which were significantly inhibited by IL-10, increased MHC II expression when IL-10 was blocked. Thus, MHC II expression in HBV+HCV-infected DCs is likely modulated by high concentrations of IL-10. However, an IL-10-independent pathway may also modulate MHC class II expression in co-infected DCs (38).
Our data suggest that the core proteins of HBV and HCV in co-infected patients synergistically stimulate DCs to overproduce IL-10. Excess levels of IL-10 inhibit production of pro-inflammatory cytokines such as TNF-α, IL-6, and IL-12 and downregulate MHC II expression. Coupled with impaired antigen presentation, a lack of pro-inflammatory cytokines causes CD4+ T-cells to be insufficiently stimulated, anergized, or tolerized. Moreover, direct viral inhibition of CD4+ T-cells and IL-10-induced anergy may pose significant barriers to immune activation (29). Unopposed viral replication may yield a significantly higher viral titer, infecting more hepatocytes and accelerating progression to hepatocellular carcinoma.
Our studies reflect the immediate and mutual effects of HBV and HCV core proteins on human DCs, which could serve an important role in blunting the early antigen priming and stimulation of T-cell responses, leading to downstream tolerization of antigen-specific T-cells and establishment of chronicity. Further studies are required to characterize the interprotein interactions and intracellular signaling pathways that lead to hepatitis virus core protein-mediated suppression of DC function.
Footnotes
Acknowledgments
This work was supported by MOP 123377 and MOP 79327 grants from Canadian Institutes of Health Research (CIHR). Dr. Mark Peppler is gratefully acknowledged for his careful review of the manuscript.
Author Disclosure Statement
No competing financial interests exist.