
Abstract
Members of the species Zaire ebolavirus cause severe hemorrhagic fever with up to a 90% mortality rate in humans. The VSVΔG/EBOV GP vaccine has provided 100% protection in the mouse, guinea pig, and nonhuman primate (NHP) models, and has also been utilized as a post-exposure therapeutic to protect mice, guinea pigs, and NHPs from a lethal challenge of Ebola virus (EBOV). EBOV infection causes rapid mortality in human and animal models, with death occurring as early as 6 days after infection, suggesting a vital role for the innate immune system to control the infection before cells of the adaptive immune system can assume control. Natural killer (NK) cells are the predominant cell of the innate immune response, which has been shown to expand with VSVΔG/EBOV GP treatment. In the current study, an in vivo mouse model of the VSVΔG/EBOV GP post-exposure treatment was used for a mouse adapted (MA)-EBOV infection, to determine the putative VSVΔG/EBOV GP-induced protective mechanism of NK cells. NK depletion studies demonstrated that mice with NK cells survive longer in a MA-EBOV infection, which is further enhanced with VSVΔG/EBOV GP treatment. NK cell mediated cytotoxicity and IFN-γ secretion was significantly higher with VSVΔG/EBOV GP treatment. Cell mediated cytotoxicity assays and perforin knockout mice experiments suggest that there are perforin-dependent and -independent mechanisms involved. Together, these data suggest that NK cells play an important role in VSVΔG/EBOV GP-induced protection of EBOV by increasing NK cytotoxicity, and IFN-γ secretion.
Introduction
E
Immunization with VSVΔG/EBOV GP protects 100% of cynomolgus macaques and rhesus macaques from a high-dose lethal challenge of EBOV (18,26). Immunizing these animals with VSVΔG/EBOV GP results in the development of low levels of EBOV-specific neutralizing antibodies, and an increased interferon (IFN) and cellular response to the EBOV infection (3,26). The VSVΔG/EBOV GP vaccine has also been utilized as a therapeutic post-exposure treatment to protect mice, guinea pigs, and NHPs from a lethal challenge of the EBOV (11,13,19). NHPs receiving the VSVΔG/EBOV GP treatment 20–30 min following EBOV challenge developed signs of HF with either no or low transient plasma viremia, and had a 50% survival rate (11,13,19). Survivors from these studies developed both IgM and IgG EBOV GP-specific antibody serum levels (11,13,19). What has become clear is that an EBOV infection causes rapid mortality in human and animal models, with death occurring as early as 6 days after infection, just when the adaptive immune response is developing. This suggests a vital role for the innate immune system to control the infection before cells of the adaptive immune system can assume control and completely clear the virus from the host. One cell type within the innate immune system that can cause rapid response to infection is the natural killer (NK) cell.
NK cells have been considered a critical first line of defence against infection as the adaptive immune system is being activated (10,45). NK cells are derived in the bone marrow and become licensed mature killers in the periphery (28,45). NK cells kill infected target cells by granule exocytosis regulated by a balance of inhibitory and activating receptors found on the cell surface (41). Target cells, once infected, change the expression of inhibitory and/or activating ligands on their surface, thus changing the balance of NK cell function. Such changes in target ligand surface expression have been observed during an EBOV infection, where inhibitory markers, such as the major histocompatibility complex (MHC) class I, decreased on the surface of infected cells (40,42). Theoretically, with the decrease in MHC class I on the target cell, NK cells may become activated to kill EBOV-infected cells. However, a study performed in guinea pig-adapted EBOV-infected baboons indicated a steady decrease in NK cell activity over 6 days, with a burst above normal on day 8 coinciding with a fever, before declining to below normal by death (24). Additionally, EBOV does not cause NK cells to secrete cytokines such as IFN-γ or tumor necrosis factor (TNF)-α during direct stimulation in vitro (43). Taken together, this suggests that NK cell activity may be compromised during an EBOV infection. This previous evidence demonstrated that NK cell function is wholly or partially abrogated during an EBOV infection, which is supported by NHP data where the percentage of NK cells in the peripheral blood mononuclear cells (PBMCs) decreased by nearly 75% 4 days after EBOV infection in cynomolgus macaques (20,38). However, when rhesus macaques were given VSVΔG/EBOV GP after an EBOV infection, there was a marked increase in NK cells in the PBMC population (13). These data, demonstrating an increase in NK numbers during the post-exposure treatment, may indicate an important role for NK cells in controlling an EBOV infection during the early time points. However, the mechanism of NK protection remains to be proven.
The importance of NK cells during an EBOV infection has been confirmed in murine models. Typically, EBOV does not cause infection in immunocompetent adult mice (12). However, by passaging the virus in suckling mice, a mouse adapted (MA) Ebola virus (MA-EBOV) does cause a lethal infection in adult mice (9). Utilizing EBOV virus-like particles (VLPs) as a vaccine platform, Warfield et al. demonstrated that wild type (WT) mice stimulated with EBOV VLPs survived a lethal challenge of MA-EBOV, whereas mice that did not have NK cells succumbed (16,43). Stimulation of NK cells with EBOV VLPs resulted in the secretion of cytokines (IFN-γ, TNF-α, IL-4, -5, -6, -12, and MIP-1α) that have been shown to play a role in controlling an EBOV infection, as well as initiating killing activity of the NK cells (4,16,43). Immunization with VSVΔG/EBOV GP also stimulated IFN-γ and TNF-α responses in infected cynomolgus macaques (26). However, cytolytic activities of NK cells has yet to be determined using the VSVΔG/EBOV GP platform.
Here, we use an in vivo mouse model of the VSVΔG/EBOV GP post-exposure treatment for a MA-EBOV infection to determine the possible protective mechanism of NK cells. To elucidate the role of NK cells in the treatment with the VSVΔG/EBOV GP virus further, we utilized a modified flow cytometric cell-mediated cytotoxic (CMC) assay to determine the ability of VSVΔG/EBOV GP to activate NK cells (21). These assays will facilitate insight into how VSVΔG/EBOV GP, used as a post-exposure therapeutic, aids animals to survive a lethal MA-EBOV challenge and further our understanding of the innate immune system's role in protection against filoviruses.
Materials and Methods
Animal studies
Six- to eight-week-old male and female Balb/C mice (Charles River), male C57Bl/6 (Taconic), or male C57Bl/6-Prf1tm1Sdz/J Perforin (Pfn−/−) deficient (Jackson Laboratory) mice were used. For the NK depletion experiment, the C57Bl/6 mice (n=5) were inoculated intraperitoneally (i.p.) with 30 μL of anti-Asialo GM1 antibody (Wako Chemicals) to deplete the NK cells, or 30 μL of anti-rabbit IgG antibody (Sigma) as a nonspecific control, every 4 days starting 9 days prior to infection and continuing throughout the experiment. Animals were challenged i.p. with either 1,000 LD50 of MA-EBOV or Dulbecco's modified Eagle's medium (DMEM), followed by treatment with 2×105 pfu of VSVΔG/EBOV GP or DMEM 24 h later. Their temperatures and weights were recorded daily for 2 weeks, but survival was followed for 28 days. For the Pfn−/− experiment, the mice were infected i.p. with either 1,000 LD50 of MA-EBOV or plain DMEM, followed by an i.p. treatment 24 h later with either 2×105 pfu VSVΔG/EBOV GP or DMEM. Survival was followed for 28 days. All animal work was performed in a BSL-4 laboratory at the Canadian Science Centre for Human and Health (CSCHAH), and was approved by the CSCHAH Animal Care Committee following the guidelines of the Canadian Council on Animal Care.
Reagents
IFN-γ or -β enzyme-linked immunosorbent assays (ELISAs; Cedarlane Laboratories) were performed on the supernatant of infected and uninfected cells as per the manufacturer's directions. EBOV GP monoclonal antibody ZGP42/3.7, generously provided by Ayato Takada (University of Tokyo, Japan) and monoclonal anti-EBOV GP 5D2 (National Microbiology Laboratory, Canada) were utilized in Western blots to detect EBOV GP. Monoclonal anti-VSV GP antibody (P5D4, Sigma-Aldrich) was used in Western blots to detect VSV G. Goat anti-mouse horseradish peroxidase (HRP) antibody (Sigma-Aldrich) was utilized as a secondary antibody for Western blots.
Cell lines
The human K562 lymphoblast cell line was cultured in RPMI 1640, 2.393 g/L HEPES, 2 mM L-glutamine (L-Glu), 1.5 g/L sodium bicarbonate, 0.11 g/L sodium pyruvate (Invitrogen), supplemented with 10% heat inactivated (HI) fetal bovine serum (FBS; Multicell Technologies), 10,000 IU/mL penicillin, and 10,000 μg/mL streptomycin (Pen/Strep; Invitrogen). NK-92MI lymphoblast cell line was cultured in Alpha Modified Minimum Essential Medium Eagle (Sigma-Aldrich), 12.5% HI FBS, 12.5% HI horse serum (Invitrogen), 0.2 mM folic acid, 0.2 mM myo-Inositol, 0.1 mM 2-Mercaptoethanol (βME), Pen/Strep, and 2 mM L-Glu. The RAW 264.7 mouse macrophage cell line was cultured in DMEM, 10% HI FBS, and Pen/Strep. All cell lines were maintained in a 37°C, 5% CO2 humidified incubator.
Viruses
VSVΔG/EBOV GP was generated as described previously (17). In brief, the gene for the GP of VSV Indiana serotype was substituted with the Zaire ebolavirus GP gene (strain Kikwit). WT VSV strain Indiana, Zaire ebolavirus strain Kikwit (EBOV), and mouse-adapted EBOV strain Mayanga (MA-EBOV) (9) were grown in Vero E6 cells (ATCC) at 37°C, 5% CO2.
Reverse transcriptase polymerase chain reaction and Western blot procedures
Cells were incubated with EBOV, VSVΔG/EBOV GP, WT VSV, or RPMI 1640 medium, 3% FBS for 24 h. The cells were harvested and resuspended in 6× gel loading buffer for a Western blot analysis, while the supernatant was collected for reverse transcriptase polymerase chain reaction (RT-PCR) analysis. For the Western blot, samples were run on a 10% SDS-PAGE gel before transfer to a PVDF membrane using a Trans-Blot SD semi-dry transfer cell. After blocking the membrane overnight in PBS 5% skim milk, 0.1% Tween-20 at 4°C, the membrane was probed with the primary antibodies, washed with PBS, 0.1% Tween-20, followed with addition of the secondary HRP antibody before adding ECL Plus substrate for visualization of the bands on Hyperfilm. The Western blot antibodies are listed in the reagents section. The RNA from the supernatant was extracted using a QIAamp® Viral RNA Mini Kit (Qiagen) according to the manufacturer's protocol. RT-PCR was performed on the RNA samples on a Biometra Thermocycler, using primers for the EBOV GP (Forward CGC TGA AGG TGT CGT TGC; Reverse CCT TGA CTG TGC ACT TGA AC) or the VSV nucleoprotein (N) (Forward TCA TTG ACA ACA CAG TCA TAG; Reverse GCT TTC AAG GAT ACA AGG TC). The resulting samples were run on a 0.8% agarose gel. Real time RT-PCR was performed using a QuantiTect Probe RT-PCR kit (Qiagen) as per the manufacturer's directions with primers designed against the EBOV GP (Forward ACT TTC GCT GAA GGT GTC GT; Reverse AAR GGG TGT GAG CTG AAG AA; probe FAM-CAT TTC TGA TAC TGC CCC AAG CTA AGA AGG A or NP (Forward TGC CGA CGA CGA GAC GT; Reverse CGT CCC TGT CCT GTT CTT CAT C; probe FAM-AGY CTT CCG CCC TTG GAG TCA GA).
Immunofluorescence assay
NK-92MI cells were incubated with either WT VSV or VSVΔG/EBOV GP at a multiplicity of infection (MOI) of 1 for 24 h. The cells were centrifuged onto a microscope slide using a cytospin cassette. The cells were fixed with a 4% paraformaldehyde (PFA) solution for 15 min RT. Then 100 μL of a cell suspension (0.5×106 cells/mL) was placed in the circle of a prewetted filter on the slide and centrifuged at 70 g for 3 min at 4°C. The microscope slide was air-dried. The cells were washed three times with PBS and incubated with a 1:2,000 dilution of the primary antibody (2GP12/1.1 for VSVΔG/EBOV GP; anti-VSV GP antibody; Sigma Aldrich) for 1 h at 37°C. The cells were washed three times with PBS, then incubated with a 1:10,000 dilution of Alexa Fluor 488 tagged secondary goat anti-mouse antibody (Invitrogen) for 1 h at 37°C. The cells were washed three times with PBS. One drop of ProLong Gold antifade reagent with DAPI (Invitrogen) was added to stain the cell nuclei. The cells were visualized under a fluorescence microscope, and an Axiovision 4 v.4 (Zeiss) program was utilized to detect and capture fluorescent images.
NK cell mediated cytotoxicity assay
Target cells, K-562 or RAW 264.7, were incubated with VSVΔG/EBOV GP or RPMI 1640 with 10% HI FBS for 24 h at 37°C, 5% CO2. The cells were washed then resuspended in plain RPMI containing 5 μM red fluorescent cell membrane dye PKH-26 (Sigma-Aldrich) and 0.3 μM 5-(and 6)-carboxyfluoresceindiacetatesuccinimidyl ester (CFDA SE) (Invitrogen) according to the manufacturer's protocol. After adding an equal volume cold FBS, cells were washed with plain RPMI and resuspended in 100 μL RPMI, 10% FBS, at a concentration of 2.5×106 cells/mL. NK-92MI cells or IL-2 stimulated mouse NK cells were suspended in 100 μL RPMI with 10% FBS at concentrations in accordance with the Effector:Target (E:T) ratio needed for the experiment. Infected or uninfected PKH-26 and CFSE co-stained target cells were added to NK effector cells at the designated E:T ratio and incubated for 4 h at 37°C, 5% CO2. The plate was centrifuged and pellets resuspended in 4% PFA before acquiring on a BD FACSCalibur using Cell Quest Pro v4 software, and analyzed using the FlowJo 7.25 software (Tree Star, Inc.). The percent cytotoxicity for each treatment of the NK cell mediated cytotoxicity assay was calculated utilizing the following equation: % Cytotoxicity=(Dead Target cells/Total Target cells)×100%. The percent cytotoxicity was calculated by subtracting the percentage without NK cells from the percentage with NK cells to remove background cell death due to the treatment.
Statistics
Statistical tests used include the log-rank test for the Kaplan–Meier Survival curves, and for all other assays, an unpaired two-tailed t-test was performed with the GraphPad Prism 4 software (v4.03; GraphPad Software, Inc.).
Results
NK cells improve survival with VSVΔG/EBOV GP post-exposure treatment of MA-EBOV-infected mice
Previous work has demonstrated that MA-EBOV-infected mice treated with VSVΔG/ZEBOV GP 30 min or 24 h after infection all survived (13). Death from EBOV generally occurs between 6 and 9 days, suggesting the innate immune response is unable to control EBOV until the B- and T-cells can take over. As the NK cells are the predominant effectors in the innate immune system, the role of NK cells during an EBOV infection, and whether VSVΔG/EBOV GP might improve survival by modulating NK cell activity, was investigated. In order to determine whether NK cells were involved, C57Bl/6 mice were depleted of NK cells with anti-Asialo GM1 antibody (NKneg) prior to being infected with MA-EBOV and then treated with VSVΔG/EBOV GP 24 h later (Fig. 1). The mice treated with anti-rabbit antibody as a nonspecific control constituted the nondepleted NK cell mice (NKpos). Spleens were taken, and the level of NK depletion was assessed by flow cytometry to be 80–95% (Supplementary Figure S1; Supplementary Data are available online at

Natural killer (NK) cells improve survival of a post-exposure VSVΔG/EBOV GP treatment of mouse adapted (MA) Ebola virus (EBOV)-infected C57Bl/6 mice. In order to determine whether NK cells had a role in improving
NKpos and NKneg mice given media only or media+VSVΔG/EBOV GP all survived, did not lose weight, and did not demonstrate a decrease in temperature (Fig. 1). NKneg mice infected with MA-EBOV and then treated with VSVΔG/EBOV GP 24 h later all lost weight and had a decrease in temperature before succumbing to the infection (Fig. 1, left panel). These mice survived longer than the NKneg mice treated with MA-EBOV then media (p=0.0027). In contrast, MA-EBOV-infected NKpos mice treated with VSVΔG/EBOV GP at 24 h had 20% survival, with the surviving mouse recovering its lost weight and temperature (Fig. 1A, right panel). A comparison of the MA-EBOV-infected NKneg (0% survival) and NKpos (20% survival) mice treated with VSVΔG/EBOV GP demonstrates that NK cells may play a role in the VSVΔG/EBOV GP post-exposure treatment of mice infected with MA-EBOV.
EBOV and VSVΔG/EBOV GP do not infect or replicate in NK-92MI cells
The ability of MA-EBOV and VSVΔG/EBOV GP to infect and replicate in NK-92MI cells was investigated in order to determine whether infection with either of these viruses may be responsible for a change in NK cell function. An immunofluorescence assay was performed to determine whether NK-92MI cells could be infected by the WT or recombinant VSV (Fig. 2A–C). VSVΔG/EBOV GP or WT VSV was added to NK-92MI cells at a MOI of 1, and then 24 h later stained for the EBOV or VSV GP. WT VSV but not VSVΔG/EBOV GP was able to infect the NK-92MI cells (Fig. 2B and C). As confirmation that VSVΔG/EBOV GP did not infect NK-92MI cells, a GP-specific Western blot and RT-PCR assay was conducted to look for viral replication. NK-92MI cells were treated with EBOV, WTVSV, or VSVΔG/EBOV GP at different MOIs for 72 h. EBOV replication was not supported in NK-92MI, as EBOV GP could not be detected in supernatant nor cell lysates of NK-92MI cells (Fig. 2D). Using real time RT-PCR, EBOV RNA was detected in the supernatant. However, the amounts of virus found in the supernatant did not increase over time at any MOI (Fig. 2G), thus inferring the lack of ability of EBOV to replicate in NK cells. VSVΔG/EBOV GP was in fact unable to infect NK-92MI cells, as determined by a lack of protein production in the supernatant and cell lysate of infected cells (Fig. 2E) and the lack of detection of viral mRNA in the supernatant of infected cells (Fig. 2H upper panel). The opposite was seen with VSV WT, as viral protein and mRNA was detected in the supernatant and cell lysates of infected cells (Fig. 2F and H lower panel). Therefore, VSVΔG/EBOV GP was not able to infect or replicate in NK-92MI cells.
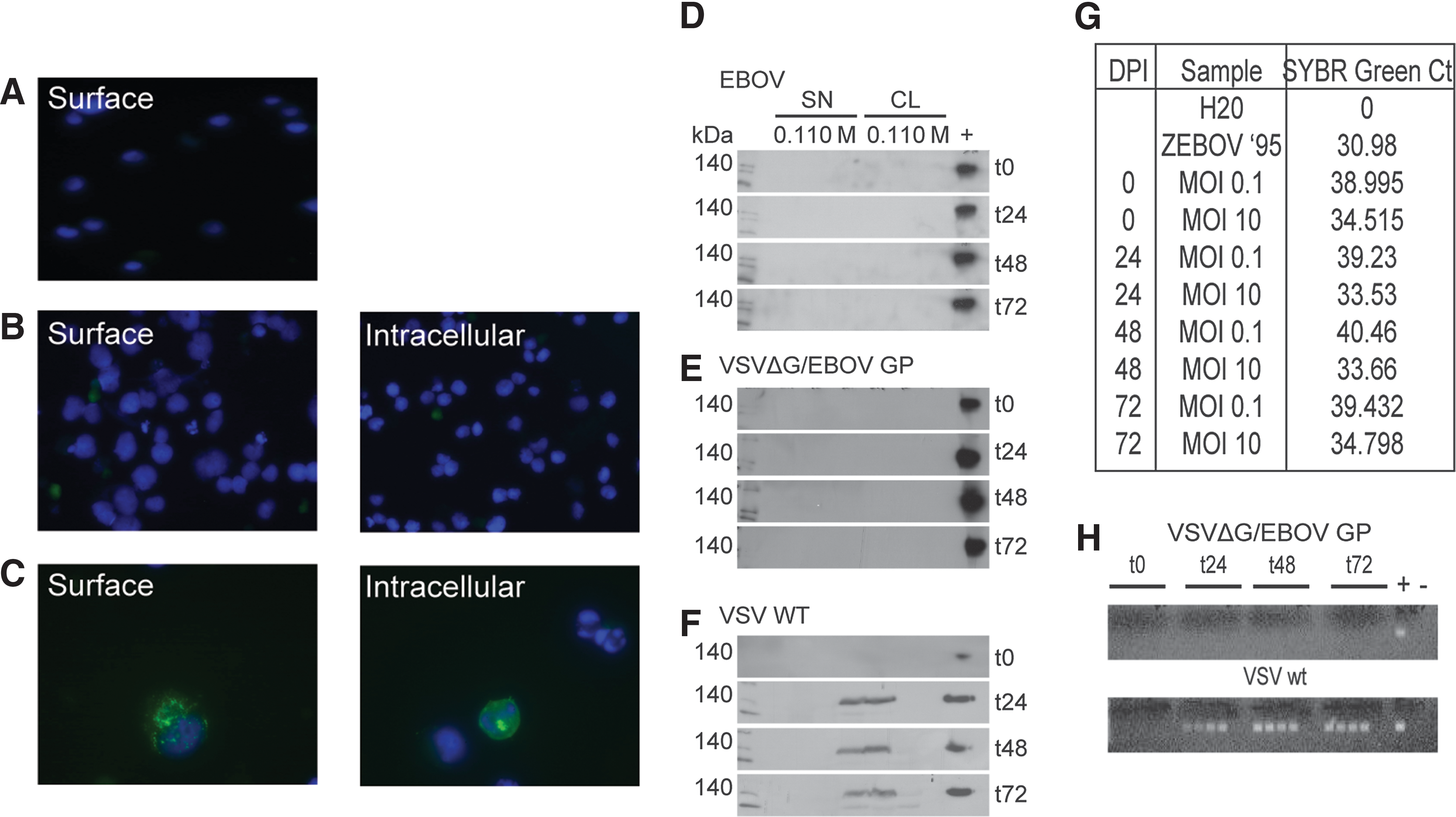
EBOV and VSVΔG/EBOV GP do not infect or replicate in NK-92MI cells. NK-92MI cells were either
Role of perforin in post-exposure treatment of MA-EBOV-infected mice with VSVΔG/EBOV GP
It has been demonstrated with EBOV VLPs as a vaccine that perforin-deficient mice do not survive a lethal challenge with MA-EBOV, while IFN-γ deficient mice do survive (43). Therefore, the role of perforin in a MA-EBOV infection in Pfn−/− mice with and without VSVΔG/EBOV GP treatment after 24 h was examined (Fig. 3). When infected with MA-EBOV, the Pfn−/− mice survived significantly longer (p=0.0183) with a MTD of 9.3 days compared to the parental WT C57Bl/6 mice (MTD=7.8 days). This suggests that perforin may partially exacerbate the disease process in MA-EBOV infections. Pfn−/− mice that received VSVΔG/EBOV GP but not MA-EBOV all survived, indicating that VSVΔG/EBOV GP is controlled in a perforin-independent manner. When the MA-EBOV infected Pfn−/− mice were treated with VSVΔG/EBOV GP, there was 50% survival with a MTD of 11 days. The MA-EBOV infected Pfn−/− mice survived significantly longer (p=0.0058) when they received VSVΔG/EBOV GP, suggesting that VSVΔG/EBOV GP treatment enhances survival in a perforin-independent manner. However, as survival was only 50%, perforin has only a partial role in survival.
A post-exposure VSVΔG/EBOV GP treatment of MA-EBOV-infected perforin-deficient mice (Pfn−/−) improves survival. The survival of four groups of mice, three groups of Pfn−/− (each n=4), and one group of C57Bl/6 (n=6) were followed for 28 days with no change in survival after 14 days. Two of the Pfn−/− groups were infected i.p. with 1,000 LD50 MA-EBOV, followed by one group receiving media (MA-EBOV/media) and the other group receiving 2×105 PFU VSVΔG/EBOV GP (MA-EBOV/VSVΔG/EBOV GP) 24 h later. The third Pfn−/− group received media in place of MA-EBOV but was treated with 2×105 pfu VSVΔG/EBOV GP (Media/VSVΔG/EBOV GP) 24 h later. The control C57Bl/6 WT mice group was infected with 1,000 LD50 MA-EBOV but not treated with VSVΔG/EBOV GP (C57Bl/6 MA-EBOV/Media).
VSVΔG/EBOV GP stimulates NK cells to secrete IFN-γ
To characterize whether VSVΔG/EBOV GP affects NK cell function, NK-92MI cells were treated with EBOV, VSV WT, or VSVΔG/EBOV GP at a MOI of 1 to determine whether these viruses stimulated the NK cells to produce IFN-γ (Fig. 4). EBOV was unable to stimulate NK-92MI cells to produce IFN-γ above background levels (∼12 pg/mL). However, VSV WT stimulated a gradual increase of IFN-γ from NK-92MI beginning at 8 h (∼24 pg/mL) and peaking at 24 h (∼130 pg/mL). VSVΔG/EBOV GP stimulated a large secretion of IFN-γ from the cells by 8 h (∼220 pg/mL) of infection, which was near the maximal peak at 24 h (∼240 pg/mL). These data demonstrate that at 8 h, VSVΔG/EBOV GP has the capacity to stimulate NK cells to secrete a burst of IFN-γ 10-fold higher than was seen with VSV WT.
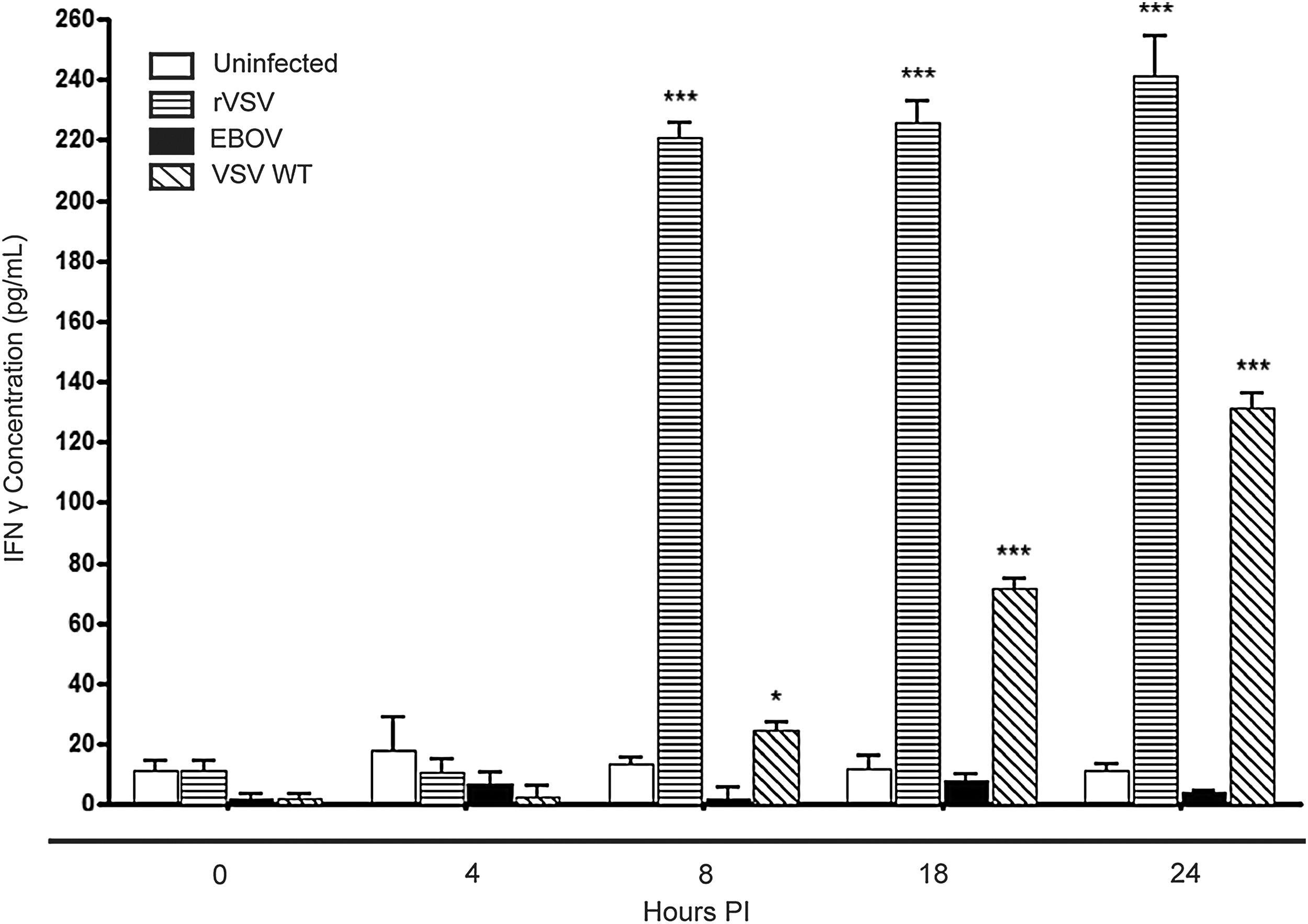
VSVΔG/EBOV GP and VSV WT stimulate interferon (IFN)-γ release from NK-92MI cells. Media, EBOV, VSV WT, or VSVΔG/EBOV GP were added to NK-92MI cells for 24 h at a MOI of 1 before removing the SN. The SN was added to an IFN-γ enzyme-linked immunosorbent assay (ELISA) in order to quantitate the amount of IFN-γ each cell produced in the presence of the various viruses. t-Tests were performed between the uninfected control samples and infected samples. *p≤0.05; ***p<0.0005. Error bars represent the standard error of the mean (SEM; n=9).
VSVΔG/EBOV GP infection of target cells causes an increase in NK cell cytotoxicity
The ability of VSVΔG/EBOV GP to stimulate NK cells to degranulate and kill infected cells was investigated in a NK cytotoxicity assay. The human lymphoblast cell line K562 and the mouse macrophage cell line RAW 264.7 were utilized as NK targets. VSVΔG/EBOV GP was able to infect and replicate in both K562 and RAW cells as determined by Western blot and RT-PCR (Fig. 5A and B). K562 cells were incubated with media or VSVΔG/EBOV GP at a MOI of 1 or 10 for 24 h. Following incubation, the cells were dually stained with PKH-26 and CFSE to distinguish between live and dead target cells, then NK-92MI cells were added for 4 h before acquiring the samples on a flow cytometer. NK-92MI had a background cytotoxicity of 12% on media treated K562 cells (Fig. 5C and D). As expected, NK-92MI had a significantly higher percentage of cytotoxicity of 25% and 35% on K562 cells infected with VSVΔG/EBOV GP at MOI of 1 and 10 respectively (Fig. 5C). At a 1:1 E:T ratio, an increase in MOI resulted in a doubling of NK cytotoxicity over background (Fig. 5C). When the E:T ratio was increased to 3:1, no difference in cytotoxicity was noted between the higher (10) and lower (1) MOIs (Fig. 5D).

VSVΔG/EBOV GP-infected target cells enhance NK-92MI cytotoxicity. A flow cytometry cell mediated cytotoxic (CMC) assay was performed to examine whether VSVΔG/EBOV GP-infected targets enhanced NK cytotoxicity.
To demonstrate this effect in a mouse model, RAW 264.7 mouse macrophage cells were incubated in media or VSVΔG/EBOV GP at a MOI of 1 for 24 h. Following incubation, the cells were also dually stained with PKH-26 and CFSE to distinguish live and dead target cells, and the cells were run on a flow cytometer subsequent to IL-2 stimulated mouse NK cell addition. The IL-2 stimulated mouse NK cells had a background cytotoxicity of 22% on uninfected RAW 264.7 cells (Fig. 5E). The mouse NK cells also had a significantly higher percentage (∼40%) of cytotoxicity on the RAW 264.7 cells infected with VSVΔG/EBOV GP (Fig. 5E). This increased cytotoxicity over uninfected cells seen in both the human and mouse NK cells demonstrated that VSVΔG/EBOV GP infection of target cells caused an increase in NK cells cytotoxicity.
In order to understand the mechanism of enhanced NK activity, the levels of classical MHC I on both target cell lines was investigated (Fig. 6). K562 cells were incubated with VSVΔG/EBOV GP, and flow cytometry was used to assess the cell surface levels of EBOV GP and MHC I at various time points (Fig. 6A and B). Both the EBOV GP and HLA increased gradually over the infection, with ∼22% of the cells expressing EBOV GP and 10% of the cells, demonstrating an increase in HLA at 24 h post-infection. However, when selecting only the EBOV GP-positive K562 cells, the percentage of cells expressing MHC I decreased from 85% to 40% by 8 h, which was sustained up to the 24 h time point. Similar results were seen for RAW 264.7 cells where the MHC I decreased as the percentage of EBOV GP-positive cells increased (Fig. 6D and E). The levels of IFN-β were also assessed at 24 h following infection. The IFN-β level secreted from K562 cells was below background but exceeded 20 pg/mL in the macrophage RAW cell line (Fig. 6F).

VSVΔG/EBOV GP infection of target cells alters major histocompatibility complex (MHC) presentation and IFN-β secretion. Expression levels of MHC I and IFN-β were assessed by flow cytometry and ELISA assays, respectively, in K562 and RAW 264.7 cells. By flow cytometry, there is an increase in expression of
Discussion
The use of VSVΔG/EBOV GP as a post-exposure therapeutic for a lethal EBOV infection in mice and NHPs results in an increase in survival when given 24 h or 30 min, respectively, after an EBOV infection (13). The physiological mechanism behind the effectiveness of VSVΔG/EBOV GP is currently unknown. However, cynomolgus macaques succumb to EBOV within 5–7 days just when rapid expansion of the adaptive immune system should begin. This suggests that the innate immune response is unable to control the virus during the early stages of infection. NK cells are the major effector in the innate immune response. Therefore, the current study focuses on the contribution of NK cells when VSVΔG/EBOV GP is used as a post-exposure treatment for a lethal MA-EBOV infection in mice.
All MA-EBOV-infected NKpos WT mice died. However, NK-depleted mice died significantly earlier, indicating that NKs have a role in combating the EBOV infection. NK cells are targeted early in EBOV infections. In EBOV-infected cynomolgus macaques, at least 66–75% of the NKs were depleted within 4 days (20,38). The importance of NK cells in the early stages of infection is confirmed with murine data where NK depletion resulted in a decrease in survival (43). In the current study, post-exposure treatment with VSVΔG/EBOV GP enhanced survival in the NKneg mice in comparison to the media-treated counterpart, suggesting a role for non-NK-mediated protection. However, in the VSVΔG/EBOV GP-treated normal mice, there was 20% survival versus 0% in the NKneg group, indicating NK cells do have a role in the VSVΔG/EBOV GP-induced innate protection. The 20% survival seen in the C57Bl/6-treated mice is in contrast to previous studies demonstrating 100% survival in female Balb/C mice (13). Although different mouse strains were used, it appears that the difference arises from the use of males in this study rather than females. A comparison of male and female Balb/C mice in the post-exposure protocol demonstrated that 40% of the males survived compared to survival of all females (Supplementary Fig. S2). This difference may reside in subtle changes in the immune response as previously demonstrated in VSV infections that female mice had lower viral titers; increased levels of Nitric Oxide Synthase (NOS) type I, II, and III expression; earlier enhanced levels of MHC II; and improved recovery than male mice (5). Other research demonstrated that female mice have a more aggressive leukocyte population residing in the naive peritoneal and pleural cavities than male mice (39). The lower levels of protection in male mice allow for more sensitivity in the assays presented in the current study.
Several mechanisms were examined to determine how VSVΔG/EBOV GP enhanced NK cell function in an EBOV infection. Perforin is a protein required for cytolytic function in both NK cells and T-cells. VSVΔG/EBOV GP by itself was well tolerated and did not cause any disease or mortality in the Pfn−/− mice. When VSVΔG/EBOV GP was used to treat MA-EBOV-infected Pfn−/− mice, survival increased to 50% over the nontreated Pfn−/− mice, suggesting protection was due to a perforin-independent mechanism. However, since survival only increased to 50%, it is possible that there is also a partial requirement for perforin in the survival of the treated mice. This is supported by the fact that the VSVΔG/EBOV GP infection of both human and mouse target cells caused NK-92MI cells to lyse infected cells more than uninfected cells—a process requiring perforin. As NK cell cytotoxicity function is based on a balance between activating and inhibiting factors, the stimulus for this enhanced capacity may be due to the decrease in MHC I demonstrated on the VSVΔG/EBOV GP-infected K562 and RAW target cells. Hence, the fact that NK cells are able to kill VSVΔG/EBOV GP-infected cells by cytolysis should be expected, since NK cells can become activated by the loss of MHC I inhibition on the surface of the cells. Additionally, it is possible that expression of the EBOV GP on the cell surface during VSVΔG/EBOV GP infection results in stimulating NK cells to produce an increased amount of IFN-γ. In fact, in this study, NK-92MI cells secreted significantly more IFN-γ in the presence of VSVΔG/EBOV GP, a process requiring signalling via the cell surface, since VSVΔG/EBOV GP did not infect or replicate in the NK-92MI cells. It has been reported that the EBOV GP on VLPs can stimulate a monocytic cell line by Toll-like receptor (TLR)-4 (35), which is also found on NK cells (31). Since NK cells have TLR4 and the Ebola GP can interact with TLR4, the NK cells may be stimulated to act upon the infected cells more than uninfected cells. Overall, it appears that enhanced NK cell cytotoxicity may be a result of NK cell activation by EBOV GP and downregulation of MHCI by VSVΔG/EBOV GP-infected cells.
The second mechanism by which VSVΔG/EBOV GP improves survival is by counteracting the host's antiviral response. EBOV has developed strategies to block the host immune response, thereby assuring rampant replication (32,37). The primary cells infected by EBOV are the major antigen presentation cells (APCs), specifically the dendritic cells (DCs) and macrophages (20). Infection of the APCs can result in functional defects such as causing a decrease in the production of cytokines from APCs, downregulation of MHC I (23,40) or apoptosis/anergy of the NK and T and B lymphocytes. EBOV proteins VP24 and VP35, which are antagonists to the interferons required to control an EBOV infection (6,22), effectively inhibit the activation of hundreds of immune response genes. Upon exposure to VSVΔG/EBOV GP early in a MA-EBOV infection, the production of IFN-β by macrophages and potent antiviral molecule IFN-γ from NK cells may be part of the protective function that counteracts the IFN block by EBOV. Although VSV has been found to be sensitive to IFN (25) and its matrix protein has the ability to suppress type 1 interferon in infected cells (1), VSV also has the property of increasing MHC I on infected cells (7), and inducing maturation and production of type 1 interferons in plasmacytoid DCs (pDC) (2). VSVΔG/EBOV GP infects and replicates in the macrophage cell line RAW. Since VSVΔG/EBOV GP contains the same GP as EBOV needed to infect macrophages and DCs, then infection of DCs by VSVΔG/EBOV GP may counteract the EBOV-induced block on DC maturation, which leads to improper signalling of T-cells leading to apoptosis.
The in vitro assays help to build a model of how the VSVΔG/EBOV GP protective mechanism may function in the in vivo mouse model. VSVΔG/EBOV GP exerts its effects by counteracting EBOV's potent antiviral mechanisms. Ultimately, this study has demonstrated that NK cells may play a role in VSVΔG/EBOV GP-induced innate protection against a MA-EBOV infection by increasing NK cell cytotoxicity and IFN-γ secretion, and by increased IFN-β secretion by APCs. As survival was only partially restored in the presence of NKpos versus NKneg mice, then there must be other innate mechanisms of protection. Since the Ebola infection can cause mortality within 8 days following clinical onset of symptoms (33), cytolytic protection during this post-exposure treatment is possibly a function of NK cells and not T-cells. As MA-EBOV-infected perforin knockout mice survived longer than their WT C57Bl/6 controls, it is possible that cytolytic mechanisms requiring perforin exacerbate the disease process perhaps through bystander apoptosis. Additionally, the Anti-Asialo GM1 antibody also depletes basophils (34). Basophils, when activated, can secrete cytokines, such as IL-4, that can activate cells of the adaptive immune system. Depletion of basophils may result in a shift in the immune response to a more Th-1 phenotype, which could result in enhanced protection by NK cells in this study. Future NK depletion studies with NK1.1 rather than Anti-Asialo would have to be conducted to determine whether the NK protection is due to NK cells alone or the combined effects of NK cells and basophils. Overall, understanding how this post-exposure treatment performs may enable us to elucidate how our immune system functions during treatment and allow proper and safe treatment of humans.
Footnotes
Acknowledgments
This work was funded by PHAC and CSSP. We wish to thank Jonathan Audet for proofreading the manuscript, and Allen Grolla for assistance with the PCR assays.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.