
Abstract
Filoviruses subvert the human immune system in part by infecting and replicating in dendritic cells (DCs). Using gene arrays, a phenotypic profile of filovirus infection in human monocyte-derived DCs was assessed. Monocytes from human donors were cultured in GM-CSF and IL-4 and were infected with Ebola virus Kikwit variant for up to 48 h. Extracted DC RNA was analyzed on SuperArray's Dendritic and Antigen Presenting Cell Oligo GEArray and compared to uninfected controls. Infected DCs exhibited increased expression of cytokine, chemokine, antiviral, and anti-apoptotic genes not seen in uninfected controls. Significant increases of intracellular antiviral and MHC I and II genes were also noted in EBOV-infected DCs. However, infected DCs failed to show any significant difference in co-stimulatory T-cell gene expression from uninfected DCs. Moreover, several chemokine genes were activated, but there was sparse expression of chemokine receptors that enabled activated DCs to home to lymph nodes. Overall, statistically significant expression of several intracellular antiviral genes was noted, which may limit viral load but fails to stop replication. EBOV gene expression profiling is of vital importance in understanding pathogenesis and devising novel therapeutic treatments such as small-molecule inhibitors.
Introduction
D
Dendritic cells (DCs) are professional antigen presenting cells of the immune system and, in immature form, reside in peripheral tissue within the respiratory tract, gastrointestinal tract, and skin (1,15,21). Functionally, immature DCs decode signals from invading microbes with pattern recognition receptors and present the pathogen's antigens to naïve T-cells within lymph nodes. Mature, activated DCs present antigen on major histocompatibility complex (MHC) molecules, express co-stimulatory or co-inhibitory signals to T-cells, and initiate antigen-specific adaptive immune responses. However, EBOV-infected DCs fail to express co-stimulatory molecules (CD40, CD80, CD86), and they induce T-cell apoptosis, secrete some cytokines but not others, and poorly induce differentiation of allogenic lymphocytes (5,9,16,17). Given the pivotal role of DCs in the immune system, there is an urgent need to understand how EBOV disrupts DC function and influences the outcome of disease.
Gene expression profiling links multiple gene expression products to specific cellular pathways. To date, transcriptional profiling has been successfully used to examine host antiviral responses to Reston virus, EBOV, and Marburg virus (MARV) infected human liver cells and peripheral blood mononuclear cells (PBMCs) from EBOV-infected nonhuman primates (13,25). Kash et al. observed key differences in interferon (IFN)-regulated gene expression profiles between Restonvirus, EBOV, and MARV (13). Gene arrays were used to elucidate human DC responses to EBOV and evidence was found of cellular antiviral gene expression, increased MHC expression and chemokine/cytokine expression, increased expression of cell survival genes, and little or no T-cell stimulatory molecule gene expression when compared to uninfected controls.
Methods
Virus propagation
ZEBOV-95 E6p3 (three passages in VeroE6 cells) stock virus (Ebola virus/Homo sapiens/COG/1995/Kikwit-E6p3), a human isolate from the 1995 outbreak in Kikwit, DRC was grown in VeroE6 cells with Eagle's minimal essential medium (EMEM; Life Technologies, Grand Island, NY) supplemented with 1% L-glutamine, 1% HEPES, 5% fetal bovine serum (FBS), 1% penicillin/streptomycin, and 0.1% gentamycin. A separate uninfected stock of VeroE6 cells was grown in tandem with infected cells for future negative controls. All viral and control stocks were subsequently clarified by centrifugation (5 min at 250 g) and stored at −80°C in several small volume cryovials for use in individual experiments. EBOV plaque assays were done as previously described (19).
Human donors and monocyte preparation
A total of 200 mL of whole blood was collected from each donor with four heparinized 60 mL syringes. Whole blood was diluted 1:2 in Dulbecco's phosphate-buffered saline (DPBS) (without calcium and magnesium chloride and supplemented with 0.4% EDTA; Sigma-Aldrich, St. Louis, MO). Diluted blood was layered over 15 mL of 96% Ficoll (supplemented with 4% PBS) in several iterations and centrifuged at 400 g for 35 min at 20°C. PBMCs were removed and washed three times in DPBS, and monocytes were purified with the human CD14 MicroBead kit (Miltenyi Biotec, Auburn, CA) according to the manufacturer's instructions. A total of 2.0×106 monocytes were cryopreserved in 90% FBS and 10% dimethyl sulfoxide (Thermo Fisher Scientific, Waltham, MA) for future flow cytometric analysis. In several cases, an insufficient number of a specific donor's DCs were obtained for all controls and treatments.
Human DC generation
Individual donor monocytes (107 cells) were placed in T25 flasks with 10 mL RPMI supplemented with 100 ng/mL GM-CSF (PeproTech, Rocky Hill, NJ), 100 ng/mL IL-4 (PeproTech), and 50 μM 2-ME (Sigma-Aldrich). Monocytes were cultured for 5 days with 50% replacement media added on days 2 and 4. On day 5, DCs were harvested by removing media from T25 flasks and adding 3 mL of warm HyClone HyQ®Tase™ in PBS containing 0.5 mM EDTA followed by intermittent mixing for 45 min. After 45 min, the HyQ®Tase™ cell suspension was added to 50 mL conical tubes and centrifuged at 300 g for 5 min. Supernatant was removed from all tubes, and cells were resuspended and combined into 20 mL of RPMI.
Flow cytometric analysis
Flow cytometric analysis was used to confirm the identity of the monocytes and immature DCs on a FACSCaliber™ flow cytometer using CellQuest™ software (BD Biosciences, San Jose, CA). Purified monocytes and presumed DCs were stained with anti-CD14 (Life Technologies), CD11c (Life Technologies), CD1a (BD Biosciences, SK9 341651, PE), CD83 (BD Biosciences, HB15e, APC), and HLA-DR (Caltag, G46-6, PE Cy5.5). Isotype control antibodies were used to confirm the absence of nonspecific antibody binding to each cell type. All antibody staining and washes were done in BD Pharmingen™ Stain Buffer (SB; DPBS with 2% [w/v] FBS and 0.09% [w/v] sodium azide; Thermo Fisher Scientific). Prior to staining, monocytes or DCs were resuspended in 1 mL SB and 100 μL of goat serum (Thermo Fisher Scientific). For staining, 1 μL of diluted antibody was added to each tube of monocytes or DCs for 15 min. After a final wash step, DCs and monocytes were resuspended in 300 μL of BD Cytofix and stored at 4°C in the dark overnight.
EBOV infection of DCs and cell harvest, extraction, and purification
A total of 1.0×106 cultured DCs from each donor were centrifuged for 5 min at 250 g and resuspended in 100 μL media. The following conditions were uniform for each experiment: DC subsets were present in uninfected media, PolyI:C (Sigma-Aldrich), and EBOV. Samples from each condition were collected at two different time points (24 h and 48 h). Depending upon the condition, 15 μg/mL PolyI:C (final concentration) and a multiplicity of infection (MOI) of 10 for EBOV was added to each donor's DC culture and incubated for 1 h, followed by centrifugation and resuspension in 1 mL RPMI. After incubation, cells were washed, and pellets were lysed with 1 mL of TRIzol® (Life Technologies) for 15 min at room temperature and stored at −80°C.
RNA was extracted according to Invitrogen's TRIzol® extraction protocol. All TRIzol® extracted samples were dried for 10 min, resuspended in 20 μL of RNase free Tris buffer, and heated at 58°C for 10 min. The samples were then stored at −80°C. After thawing, RNA concentration was determined by NanoDrop 2000 (Thermo Fisher Scientific) and purity by Agilent RNA 6000 Nano Complete Kit with Reagents and Chips PN (Agilent Technologies, Santa Clara, CA). Each RNA sample was labeled with TrueLabeling-AMP™ 2.0 Kit (Qiagen, Valencia, CA) and Biotin-11-UTP (PerkinElmer, Waltham, MA). Labeled RNA was purified with the RNeasy kit Mini-Elute (Qiagen) protocol starting with adding 350 μL buffer RLT. The samples were eluted with 42 μL of RNase free water (Life Technologies) and examined on the NanoDrop to determine concentration.
Microarray data analysis
Gene expression products from each sample were hybridized to SuperArray Biosciences Hybplate in Hybridization Solution (Qiagen) for 18 h. RNA samples were run on SuperArray Bioscience's pathway specific human Dendritic and Antigen Presenting Cell Oligo GEArray where chemiluminescence of hybridized expression products was observed (Chemiluminescent Detection kit) on a CCD camera (LAS-3000, Fujifilm). All raw images were saved for data extraction and analysis with GEArray Expression Analysis Suite (Qiagen) software.
The raw data (spot intensity) were obtained from the GEArray Expression Analysis Suite (Qiagen). Each array, used for each donor and experimental condition, contained three spots for GAPDH. Although GAPDH gene expression varies considerably between tissues, its expression level is comparable within the same tissue across different healthy donors (2). The mean GAPDH was calculated for each array and used for data normalization by generating gene expression ratios for each gene in the array. Gene to mean GAPDH ratios were compared from array to array or, in the case of an experimental condition, from donor to donor. Multiple t-tests were used to compare gene to GAPDH ratios for 24 h EBOV-infected DCs (n=8) to 24 h uninfected DCs (n=10) and 24 h PolyI:C stimulated DCs (n=9) and for 48 h EBOV-infected DCs (n=3) to 48 h uninfected DCs (n=3) and 48 h PolyI:C stimulated DCs (n=3). Cross-comparisons between 24 and 48 h EBOV-infected and PolyI:C stimulated cultures were done with the same methodology. Results were considered statistically significant with p-values of ≤0.05, and all statistical data were evaluated on GraphPad Prism v6.01.
Results
Confirmation of DC phenotype after differentiation
DCs were analyzed for mature phenotypic markers (CD11c) and extracted RNA's stability/integrity was confirmed for quality control. All DCs used in this study demonstrated increased expression of CD11c (Table 1), and all extracted RNA was tested for degradation with the Agilent RNA 6000 Nano kit where two ribosomal peaks for each processed sample were required for further analysis (data not shown). The donor DCs presented in this study all contained markers characteristic of DCs and yielded high-quality extracted RNA for gene expression analysis.
Indicates treatment of DCs where D=done and ND=not done.
DCs were confirmed CD11c+ (≥90%) and positive for EBOV proteins by flow cytometry.
DC, dendritic cell; EBOV, Ebola virus.
EBOV-induced gene expression 24 h post-infection
GAPDH normalized gene expression data were compared among groups (uninfected 24 and 48 h vs. infected and PolyI:C 24 and 48 h). Although MOI was the same among donor DCs, viabilities over time and steady state expression levels between donors were variable. Therefore, comparing spot density for each gene from donor to donor could lead to erroneous results. EBOV-infected DCs had higher expression levels for several genes compared to cognate uninfected DCs after 24 h. Respective mean GAPDH normalized data (n=8 donors) and standard errors of 25 EBOV upregulated host genes are shown in Figure 1A, and the same normalized gene expression data are compared to normalized data of PolyI:C stimulated DCs in Figure 1B. In addition, a summary of significant gene expression differences (p≤0.05) between uninfected DCs and 24 h of EBOV infection and PolyI:C stimulation are displayed in Table 2 where individual genes are broken down into functional categories. Neither EBOV infection nor PolyI:C stimulation resulted in significant reduction in DC gene expression at 24 h. With respect to GAPDH normalized data, the following gene order represents high to low gene expression after 24 h of EBOV infection, which is also displayed in Figure 1A and B: CD83 (2.4), IL-8 (2.4), PNRC1 (2.2), HLA-F (1.8), CCL3L1 (1.8), BTG1 (1.6), ISG15 (1.2), RELB (1.1), GPX4 (0.71) ARID5A (0.70), NFκB2 (0.69), IFIT1 (0.64), SOD2 (0.59), MARCKSL1 (0.51), PIM2 (0.44), CXCL2 (0.40), CCL5 (0.27), CST7 (0.26), ICAM-1 (0.22), LAMP-3 (0.22), CXCL9 (0.22), ISG20 (0.19), EBI3 (0.18), ERBB2 (0.14), and CXCL10 (0.097).
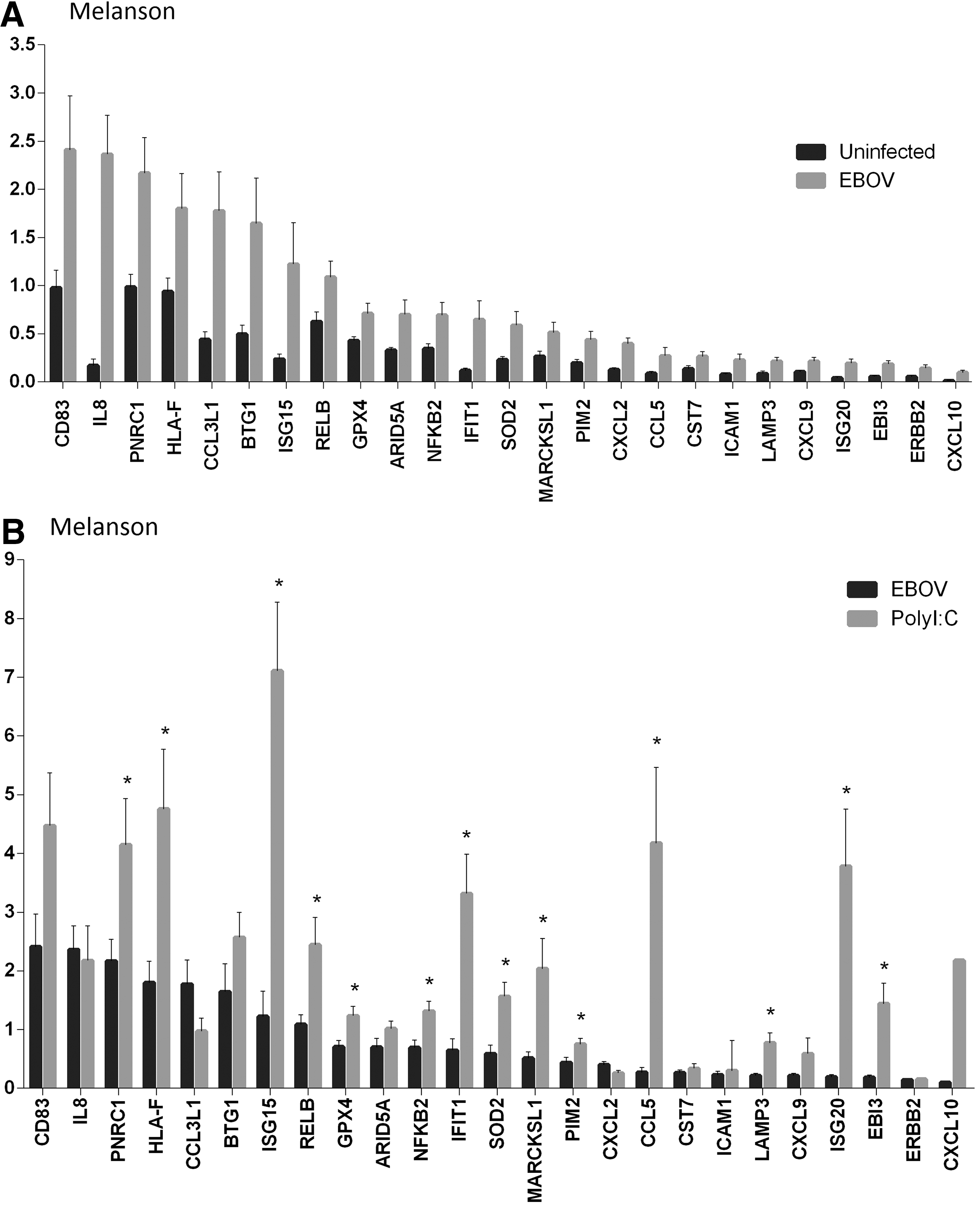
Bold indicates significant decrease from uninfected controls (p≤0.05).
Indicates significant increase from uninfected controls (p≤0.05).
As a comparison, PolyI:C stimulated human donor DCs (n=9) were evaluated for gene expression, and statistically significant gene expression changes after 24 h of stimulation are shown in Table 2. In most cases, stimulation with PolyI:C resulted in higher levels of gene expression than EBOV infection. Of the 25 significant increases in gene expression following 24 h of EBOV infection, 14 of the same genes had higher (p≤0.05) expression levels with 24 h of PolyI:C stimulation (Fig. 1B).
EBOV-induced gene expression 48 h post-infection
After 48 h, EBOV-infected DCs (n=3) had significant increases (p≤0.05) in expression of 48 genes compared to uninfected controls. Figure 2A and B and Table 3 show 48 h EBOV-infected and 48 h uninfected DC gene expression results, which break genes down into functional categories. Other than IL-18, there was no significant downregulation of genes after 48 h of EBOV infection. The following genes, ranked from high to low, based on GAPDH normalization, were shown to be upregulated after 48 h (n=3 donors) of infection: HLA-DRB5 (6.6), ACTB (6.4), HLA-DRA (6.4), IFI30 (6.4), CD68 (5.0), 18SrRNA (3.49), ISG15 (2.8), ECGF1 (2.6), CCL3L1 (2.5), CD83 (2.1), HLA-A (2.1), PNRC1 (1.9), IFIT1 (1.7), TAPBP (1.5), BTG1 (1.3), RELB (1.1), IFI35 (1.1), NFκB1 (0.98), IFI6 (0.80), IFIT2 (0.93), GPX4 (0.78), CXCR4 (0.71), SOD2 (0.69), CXCL2 (0.69), HSP90B1 (0.67), BASP1 (0.66), CCR5 (0.64), IFI44 (0.64), ARID5A (0.61), PFN1 (0.59), NFκB2 (0.54), ADAR (0.53), ISG20 (0.45), IFI27 (0.44), CCL5 (0.44), LYN (0.43), CCL8 (0.39), CXCL10 (0.37), TNF (0.36), DCTN2 (0.34), ATM (0.32), CD44 (0.32), CXCL9 (0.29), MARCKSL1 (0.28), CCL11 (0.26), IL26 (0.25), IGSF6 (0.23), and LAMP3 (0.1).
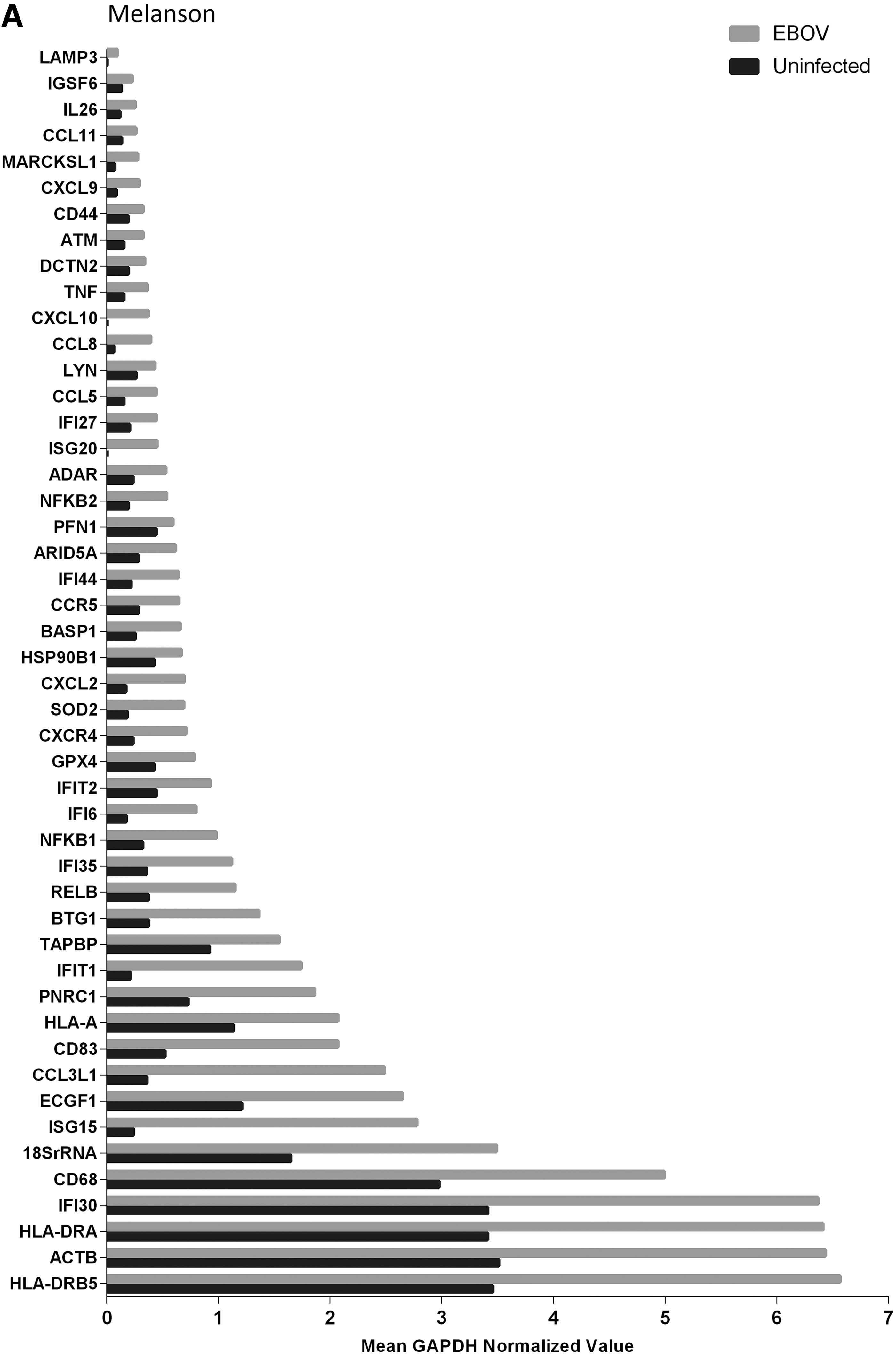

Bold indicates significant decrease from uninfected controls (p≤0.05).
Indicates significant increase from uninfected controls (p≤0.05).
As a comparison, PolyI:C stimulated human donor DCs (n=9) were evaluated for gene expression, and statistically significant gene expression changes after 48 h of stimulation are shown in Table 3. Eighty-seven genes had significant (p≤0.05) expression changes between PolyI:C stimulated uninfected DCs (Table 3). Out of the 48 genes showing expression changes in EBOV-infected DCs, 26 had significantly higher (p≤0.05) levels in PolyI:C stimulated cultures (Fig. 2B).
At both 24 and 48 h post-EBOV DC infection, 14 genes were expressed significantly higher than uninfected controls (Fig. 3). When comparing gene expression data between 24 (n=8) and 48 h (n=3) EBOV-infected DCs, significant increases (p≤0.05) in ISG20, IFIT1, and CXCL10 gene expression were seen (Fig. 3).
Persistently upregulated DC genes at 24 and 48 h post-EBOV infection. EBOV-stimulated DC gene expression shown to be statistically different from uninfected controls at 24 h were compared to the same genes that were statistically different from 48 h uninfected controls. Statistically different gene expression levels between EBOV-infected DCs at 24 versus 48 h are shown by asterisks.
Discussion
DCs facilitate early immune responses and mold acquired immunity to pathogens. Unfortunately, DCs are permissive to EBOV infection and replication and survive for several days after initial infection in culture. Furthermore, information on antiviral responses initiated by DCs is indistinct (14). Using gene arrays and human donors' DCs, gene expression was evaluated up to 48 h of in vitro EBOV infection. Consequently, DCs demonstrated intracellular antiviral responses and expressed chemokine and cytokine genes with sparse chemokine receptors and no co-stimulatory marker gene transcriptional changes from uninfected DCs.
Since DCs survive for several days after initial infection, this study has evaluated late post-infection biomarker expression. After 24 h of infection, several cytokine and chemokine transcriptional changes were noted in EBOV-infected DCs. Particularly, IL-8 expression was nearly 15 times higher than uninfected DCs (Fig. 1A). IL-8 levels become elevated in the blood of EBOV-infected patients, and infected antigen presenting cells may be responsible for IL-8 induced neutrophilia seen later in infection (8,12). In addition, CXCL2 (MIP-2α), CCL5 (RANTES), and ISG15, which are chemotatic for neutrophils, are significantly elevated after 24 h of EBOV infection (Fig. 1A), and CXCL10 (IP-10) and CXCL9 (MIG), whose expression is increased 48 h post-infection, are chemoattractant for T-cells and natural killer cells (Fig. 2A). At the gene expression level, DCs appear in a state of activation and attempting to recruit effector cells, but they fail to show significant changes in CCR7 (24 and 48 h) expression (included as a marker in the array) and may not have the capacity to home to lymph nodes. In fact, the only two receptors showing significant transcriptional changes through 48 h were CCR5 (receptor for CCL3L1 and CCL11) and CXCR4 (receptor for SDF-1).
Cellular antiviral responses to EBOV infection included expression of single strand RNase (ISG20 at 24 and 48 h post-infection) and translation repression with IFIT1 (seen at 24 and 48 h post-infection; Fig. 3). IFIT1 binds to eIF3 and subsequently suppresses >60% of translation (26). IFIT1 may reduce viral protein expression but also hinder cellular signaling and antigen presentation. Transcriptional changes in IFIT2, IFI35, ISG15, and ADAR were seen after 48 h, whose functions include disassociating double-stranded RNA and inhibition of protein translation and cell trafficking. ISG15, for example, has been shown to inhibit EBOV VP40 budding (20).
Some transcriptional changes may help EBOV by enhancing viral replication. For instance, PNRC, a nucleolar protein, facilitates rRNA synthesis, and overexpression may help EBOV synthesize viral proteins (24). This also coincides with elevated 18SrRNA after 48 h of infection (Fig. 2A). BTG1 expression has been shown to be anti-apoptotic and may allow EBOV to persist in DCs while obverting cell-programmed death (14). PIM2, NFkB1, NFkB2, and ERBB2 expression, which were increased during infection, have also been shown to promote cell survival (Figs. 1A and 2A) (3). Consequently, DCs persist while viral replication ensues. The majority of these genes, however, are increased after 24 h while several antigrowth and pro-apoptotic genes (IFIT44, IFI6, and IFI27) are expressed after 48 h, which may be in response to cellular stress or cytokines (11).
EBOV-infected DCs demonstrate maturity by significant expression of CD83 at 24 and 48 h, and expression levels are not significantly different from PolyI:C stimulated levels (Fig. 3). Furthermore, transcriptional changes in HLA-A (MHC I), HLA-DRA (MHC II alpha chain), TAPBP (tapasin), LAMP-3 and IFI30 (MHC II peptide loading), and HLA-DRB5 (MHC II beta chain) are indicative of antigen presentation (Tables 1 and 2). All of these transcriptional changes indicate normal antigen processing and presentation pathways are being initiated at the gene expression level. However, protein translation may be inhibited by ant-viral cellular defenses or the virus itself, and absence of co-stimulatory markers CD40 and/or CD80/86, which has also been observed by others at the protein expression level, is indicative that DCs may not be functionally capable of directing an acquired immune response (16).
EBOV-infected DCs demonstrated transcriptional changes among antiviral cellular genes, cell survival, MHC I and II related genes, and multiple chemoattractant genes for granulocytes and lymphocytes. However, there was no significant changes for lymph node homing receptors or T-cell co-stimulatory molecule genes. During this study, cells were stimulated with PolyI:C as a positive control, and there was robust expression of multiple genes. However, there is no way to equivocate virus MOI to PolyI:C concentration where the latter may be equivalent to hundreds of MOIs. Although gene expression does not necessarily imply protein translation, lack of expression of essential genes clearly denotes lack of protein synthesis. Furthermore, the function of several genes, presented here, in the context of EBOV infection in DCs are not known, and autocrine and paracrine effects of virus-induced mediator synthesis are not known. Overall, lack of co-stimulatory and lymph node homing receptor gene transcriptional changes in response to EBOV indicates DCs have difficulty homing to lymph nodes and activating T-cells but still may have the ability to recruit inflammatory cells to sites of infection. Clearly, several markers stand out among EBOV infection, and this visibility in a specific cell type has comparative utility in small molecule drug testing or early host diagnostic gene profiling. Such profiling would require larger sample sizes for greater statistical power and biomarker equivalence studies in clinically relevant samples.
Footnotes
Acknowledgments
The use of human subjects for research purposes is regulated by the federal government and individual institutions. The research in this study has complied with all relevant federal guidelines and institutional policies. All donor samples were collected under the human use protocol USAMRIID FY07-12. A special thanks to Dr. Janice Rusnak and RN Denise Clizbe for their assistance on this protocol, and to Dr. William Pratt for helping this project get started. This work was funded under USAMRIID FY07-09 U.S. Army In-house Laboratory Independent Research Grant. Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the United States Army, Department of Defense, or the United States Government.
Author Disclosure Statement
No competing financial interests exist.