
Abstract
In hepatitis B virus (HBV) infection, the immune reaction is responsible for viral clearance and preventing their spread within the host. However, the immune system is dysfunctional in patients with chronic HBV infection, leading to an inadequate immune response against the virus. A major factor contributing to inefficient immune function is the phenomenon of immune exhaustion. Hence, understanding immune activation and exhaustion during HBV infection is important, as it would provide insight in developing immunotherapy to control chronic HBV infection. The aim of this review is to highlight the existing information on immune effector functions and immune exhaustion in response to HBV infection.
Introduction
H
Immune cells play an important role in control against viral diseases. Upon viral antigenic challenge, T cells undergo a program of activation with subsequent clearance of infection and formation of stable memory population. The significance of T cells in controlling viral infections has been well documented in both mice and humans (2,5,67). However, persistent exposure to excessive viral antigens, such as in the case of chronic HBV infection, leads to progressive exhaustion of immune cells, which results in poor effector function (28,66,69). The diminished effector function of immune cells (immune exhaustion) is a major hurdle in the controlling and clearance of the virus in chronic infections. This review provides an overview of the immune mechanism in response to HBV, and also emphasizes the current understanding of immune exhaustion, as well as therapeutic interventions that may be considered in the future to overcome immune dysfunction in chronic hepatitis B infection.
Immune Response in HBV Infection
Both innate and adaptive immune responses are involved in HBV clearance during acute infection. Initially, the innate immune system recognizes viral pathogens through pattern recognition receptors (PRRs), including Toll-like receptors (TLRs), retinoic acid inducible gene I (RIG-I), NOD-like receptors (NLRs), C-type lectins, and others (7,8). Among the PRRs, TLRs are the most common groups, which are expressed in many types of cell, including hepatocytes and cells of the innate and adaptive immune systems. The binding of TLRs to pathogen-associated molecular pattern (PAMP) results in immediate effector functions of innate immune cells. TLRs have different subcellular localization, and can be categorized into two groups. The first group includes TLR1, TLR 2, TLR 4, TLR 5, TLR 6, TLR 11, and TLR 12, which are present at plasma membrane, while the second group comprises TLR3, TLR 7, TLR 8, and TLR 9, which are localized to the intracellular compartment such as endosomes (1).
The binding of PAMP to TLRs activates adapter proteins such as myeloid differentiation primary lymphoid protein 88 (MYD88), Toll/IL1 receptor domain-containing adaptor protein (TIRAP), Toll/IL1 receptor domain-containing adaptor protein IFN-β (TRIF), and others. These adapter proteins along with cytoplasmic domain of TLRs in turn trigger signaling molecules including the nuclear factor-κB, mitogen-activated protein kinases, interferon regulating factor-1 and 7, leading to the secretion of type I interferons and proinflammatory cytokines (52,73). Production of type I interferons such as IFN-α and IFN-β in response to PAMP induces expression of antiviral proteins. The IFN-induced antiviral proteins are 2′,5′-oligoadenylate synthetase (OAS) and RNase L, which mediate RNA degradation; the Mx protein GTPases family, which targets viral nucleocapsid and inhibits RNA synthesis; protein kinase R (PKR), which inhibits translation initiation by phosphorylating eIF-2α; and the RNA-specific adenosine deaminase (ADAR), which deaminates adenosine from double-stranded RNA to yield inosine (50). The ligands for TLR3, TLR4, TLR5, TLR7, and TLR9 have been shown to inhibit HBV replication by inducing IFN-α/β in the liver of transgenic mice (29). In addition, the liver of IFN-α/β knockout and IFN-γ knockout mice has much higher levels of HBV replicates than those of heterozygotes littermates (29,41).
Apart from a direct inhibitory effect, innate immune cells release a plethora of cytokines and chemokines, thereby amplifying the host adaptive immune system. HBV inhibitory effects of TLR and other PRRs such as RIG-I are primarily mediated by intracellular antiviral pathways through overexpression of the three PRR adaptors interferon promoter stimulator 1 (IPS-1), TRIF, and MyD88, resulting in the induction of proinflammatory cytokines (24). The innate immune system is responsible for early HBV control by noncytolytic mechanisms. However, HBV can counteract the TLR-mediated antiviral response of the innate immune system and establish itself in infected patients. This can be characterized by the presence of HBeAg in the sera of infected individuals (26). It has been demonstrated that exposure to HBeAg can downregulate TLR2 expression that results in an nonresponsive effect of TLR2 ligands (64). HBsAg, HBeAg, HBV virions, or HBV-bearing supernatants are also able to inhibit TLR-induced antiviral activity by suppressing IFN-β production and subsequent induction of interferon-stimulated gene (ISG) and nuclear factor kappa B (NF-κB) (74).
Reports have shown that TLR signaling molecules are significantly downregulated in the peripheral blood mononuclear cells of chronic HBV-infected patients, suggesting the significance of this pathway in the clearance and control of HBV load (42). Another strategy used by HBV is the use of covalently closed circular DNA (cccDNA) as a transcriptional template. This cccDNA is sequestered within the nucleus of infected hepatocytes, allowing HBV to evade innate immune recognition and replicate in the liver for years (13). In vivo studies on chimpanzees and woodchuck have demonstrated that immune response–related inducible genes were absent during entry and expansion of HBV, suggesting the absence of an innate immune response (15,71). Thus, in these models, HBV acts like a stealth virus, escaping early detection and clearance from the host.
Above all, the adaptive immune system plays a crucial role in controlling HBV infection. Although humoral immune responses such as virus-specific neutralizing antibodies exist and arise relatively late after HBV exposure, they do not seem to contribute to the early phase of viral clearance (10). Despite this, it is becoming increasingly clear that CD4+ and CD8+ T cell–mediated immune responses determine the outcome of HBV. Production of antiviral cytokines such as IFN-γ and TNF-α by CD4+ and CD8+ T cells as well as natural killer cells can inhibit the replication of the virus in hepatic cells (16,20,23,32). Both IFN-γ and TNF-α are known to be involved in the post-transcriptional control of HBV through the disruption of La proteins, HBV-RNA stabilizing factor (47,61). In the early phase of HBV infection, the CD4+ T cell is primed to activate professional antigen presenting cells (APCs) necessary for the induction of efficient antiviral CD8+ cytotoxic T cell responses (49).
The involvement of CD8+ T cells in the control of HBV infection has been demonstrated by depleting the cell population with monoclonal antibodies (58). In a transgenic mouse model, it was demonstrated that cytokines such as IFN-γ and TNF-α secreted by CD8+ T cells eliminate HBV nucleocapsid particles and also destabilize the viral RNA (21). In a chimpanzee model of HBV infection, it was shown that adoptive transfer of HBsAg-specific cytotoxic T lymphocytes (CTLs) results in recovery of the host from acute HBV infection. Intrahepatic chemokines as well as activated platelets facilitate recruitment of cytotoxic T lymphocytes, leading to the killing of HBV-infected hepatocytes (13,27,57). It was also confirmed in the chimpanzee infection model that noncytopathic T cell effector mechanism results in recovery from acute HBV infection, while cytopathic T cell effector machinery is required to remove the virus by destroying infected hepatocytes (22,55). Thus, the cell-based immune response plays a key role in the eradication of HBV from the host. However, cooperation between different kinds of cells of the immune system is critical in eradicating HBV from the host. Figure 1A shows how cells of the immune system work to tackle the virus-infected cells.
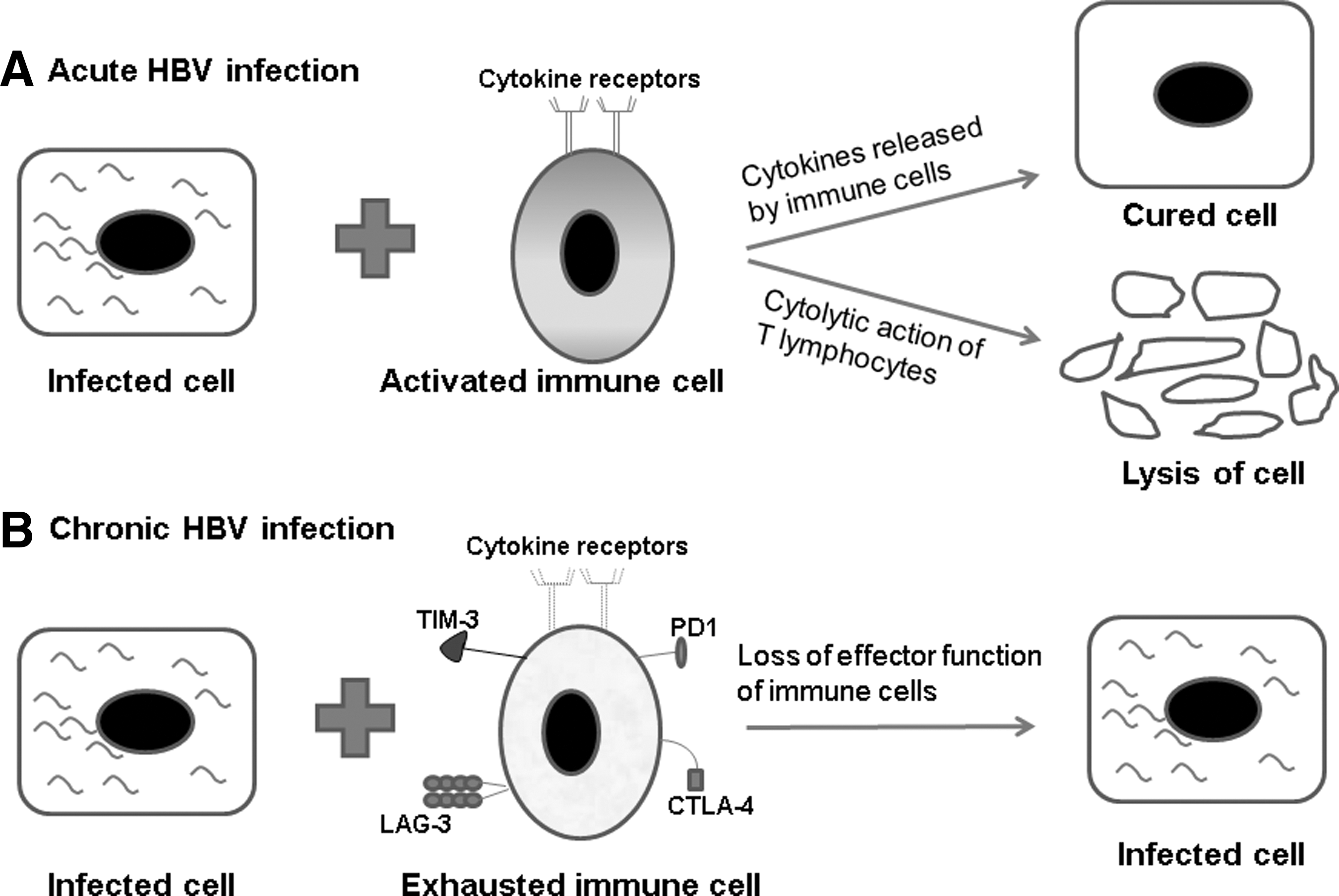
Anti-hepatitis B virus (HBV) immune responses and immune exhaustion.
Immune Exhaustion in Hepatitis B
The combined effort of innate and adaptive immune cells can decrease the viral load of infected cells (Fig. 1A). However, a key aspect of immune cell function can be substantially compromised under settings of persistent antigen stimulation (17,35,44,68). This kind of impairment in the immune cell that occurs as a result of chronic exposure to excessive viral antigens is termed “immune exhaustion.” The characteristic properties of exhausted cells include reduced proliferative potential, diminished cytokine production, and decreased cytotoxicity. The key mediator of T cell exhaustion is the upregulation of several inhibitory receptors such as cytotoxic T-lymphocyte-associated protein-4 (CTLA-4), programed-death1 (PD1), T-cell immunoglobin mucin-3 (TIM-3), and lymphocyte activation gene-3 (LAG-3; Fig. 1B). During chronic viral infections, exhausted immune cells undergo sequential phenotypic changes that results in progressive loss of effector function (11,68,70). Such functional impairment of responding T cells has been confirmed in human chronic viral infections such as HBV (56,68,70). The T cell is indispensable for the initiation and regulation of the immune response to antigen. Both the engagement of T cell receptor (TCR) with antigen-bound major histocompatibility complex (MHC) and a costimulatory signal are needed for the complete activation of T cells. The binding between CD28 receptor expressed on T cells and B7 ligands (B71 and B72) from APCs provide one of the dominant costimulatory signals. The presence of costimulatory signals such as CD28 pathway lowers the threshold for activation, enhances CD25 expression, promotes cell cycle progression, and improves survival by upregulating anti-apoptotic protein (19). Unfortunately, increased expression of inhibitory receptors such as CTLA-4, PD1, TIM-3, and LAG-3 during exhaustion suppresses the activation of T cells. The inhibitory receptor-CTLA-4 expressed on T lymphocytes has a higher affinity for B7 ligands than for CD28 (37,76). The binding between CTLA-4 and B71 or B72 ligand inhibits T cell functions, making it a powerful negative regulator of CD-28-dependent T cell responses (46).
PD1 is another inhibitory receptor of the CD28 superfamily. PD1 has been identified to be highly expressed on exhausted T cells during chronic viral infections and cancer. Signaling through the PD1 receptor is responsible for exhaustion of CD4+ and CD8+ T cells (3,12). Amplified inflammatory cytokines have been linked to upregulation of PDL1 on mDCs during chronic HBV infection, which may in turn mediate T cell exhaustion in chronic HBV-infected individuals (9). In chronic hepatitis B, there is also increased expression of TIM-3 and LAG-3, which act as negative regulators of immune activation (36,45). The downregulation of cytokine receptors such as IL-7Rα and IL-15Rα has also been associated with exhausted T cells. Lower levels of IL-7Rα and IL-15Rα can lead to defective cytokine signaling and consequently impaired T cell function (59). At a molecular level, T cell exhaustion reveals altered expression of transcription factors and changes in signal transduction (31,63). In addition, a suppressive environment created by increased Tregs also augments immune exhaustion. Collectively, these indicate that an increase in multiple inhibitory receptors and downregulation of certain cytokine receptors during chronic viral infection such as HBV results in immune exhaustion (Fig. 1B) (62). Mixed infections of HBV and HCV are common, which can further deteriorate the immune scenario (4,72). Increased levels of ligand for PD1 (PDL1) has been reported on kupffer cells, liver sinusoidal endothelial cells, and leukocytes of patients with chronically inflamed livers such as HBV and HCV (33). Additionally, high levels of PD1 on HBV- and HCV-specific CD8+ T cells have been reported during chronic infection, which may contribute to immune exhaustion and disease severity (14,18). Thus, combined modulation of inhibitory pathways in chronic HBV and HCV co-infection might be responsible for a worse disease outcome compared with HBV or HCV infection alone.
Targeting Immune Exhaustion in Clinic
Although initial observations of immune exhaustion have come from studies in lymphocytic choriomeningitis virus (LCMV), therapeutic interventions to control immune exhaustion as a potent rejuvenator of immune cells have been reported in cancer treatment (34,51,54,75). The U.S. Food and Drug Administration (FDA) have approved three monoclonal antibodies to block exhaustion markers for the treatment of metastatic melanoma. The monoclonal antibody against CTLA-4 (ipilimumab) was approved in 2011, and two monoclonal antibodies against PD1 (nivolumab and pembrolizumab) were approved in 2014. In view of the results obtained so far with the monoclonal antibodies against CTLA-4 and PD1 receptors, there appears to be a possibility of considering markers of immune exhaustion as a promising modality of immunotherapy for chronic hepatitis B patients. The mechanistic basis for the development of T cell exhaustion in chronic viral models has begun to take shape over the past few years. The ability of HBV-specific T cells to proliferate and to secrete cytokines in samples from chronic HBV individuals can be enhanced by blocking the PD1/PDL1 interaction by PDL1 antibodies (6). Similarly, preventing PD1/PDL1 interaction by an anti-PD1 monoclonal antibody results in clearance of HBV in a mouse model (62). Accordingly, it suggests that reversal of T cell exhaustion by therapeutic targeting of the immune checkpoints can control chronic HBV infection.
During co-infection of HBV with HCV or HIV infection, increased liver-related mortality has been reported. Therefore, vaccination for HBV is highly recommended. However, the HBV vaccine responsiveness can be affected by HCV- or HIV-mediated immune exhaustion. In case of HCV infection, the expression of PD1 on CD4+ T cells was relatively higher in HBV vaccine nonresponders compared with responders. Further blocking of PD1 pathway in these vaccine nonresponders improved T cell activation to ex vivo HBsAg (43). Another marker of immune exhaustion, Tim-3, was shown to be overexpressed on monocytes from HCV-infected individuals. Ex vivo blocking of the Tim-3 pathway can increase the efficacy of HBV vaccine in vaccine nonresponders (65). Similarly, immune exhaustion during HIV infection decreases the efficacy of the antiviral immune response (12,48,60). The rate of response to both intradermal and intramuscular administration of HBV vaccine has been found to be reduced in HIV-infected individuals (53). Thus, targeting immune exhaustion along with effective HBV vaccination could be a potential tool to restore active immune response against chronic viral diseases.
The outcome of HBV infection is determined by host immune responses. Thus, a strong and long-term immune response to HBV antigens is essential for the effective clearance of the virus. Nevertheless, safety concern must be taken into consideration while using immune checkpoint therapies such as anti CTLA-4 or anti-PD1. During chronic infection, HBV presumably infects all the hepatocytes, and subsequently, liver flares may arise as a result of restoration of exhausted immune cells. Hence, initial reduction of viral load by using combination therapy with other antiviral therapies such as pegylated IFN-α and nucleot(s)ide analogues before targeting immune checkpoint may be necessary to prevent the potential side effect. A recent study with a woodchuck model showed that when chronically hepadna viral-infected animals received a combination therapy of nucleoside analogue entecavir along with therapeutic vaccination and PDL1 blockade, the effector functions of CD8+ T cells were revived (38). Consistent with this observation, targeting immune exhaustion can complement the conventional strategies and will form the basis of an ideal HBV therapy. Thus, further investigation of immune exhaustion in the context of hepatitis B treatment is warranted.
Conclusions
In summary, the recognition of HBV antigen by the innate immune system results in the activation of cellular immune responses that efficiently clear viral load during acute hepatitis B infection. In addition to instigating adaptive immune responses, the cytokines released by the innate immune cells can directly act on HBV-infected cells. Nevertheless, the cellular branch of adaptive immune cells acts as a major defense mechanism to combat HBV pathogenesis. On the other hand, persistent antigenic challenge makes the immune cells exhausted, resulting in their diminished cytokine production, inefficient cytolytic potential, and decreased proliferative response. Consequently, the virus persists in the host and establishes chronic HBV disease. The immunotherapy-based treatment of chronic HBV infection using monoclonal antibodies against the marker of exhaustion can be done, but further in-depth studies are required to explore the best protocols to use them in combination with antiviral drugs.
Footnotes
Acknowledgments
We thank Asif A. Dar and Rushikesh Patil for critically reading and editing the manuscript. Dimpu Gogoi is supported by INSPIRE Faculty grant from the Department of Science and Technology, India.
Author Disclosure Statement
No competing financial interests exist.