
Abstract
The Epstein–Barr virus (EBV) transactivator protein (ZEBRA) is an immediate–early protein that plays an important role in the switch from latency to productive cycle in EBV virus. ZEBRA is an important marker of EBV reactivation. In order to diagnose EBV infection status correctly and timely, a novel immunoassay was developed based on an indirect time-resolved fluoroimmunoassay (TRFIA) for Zta IgA, which used recombinant Zta antigen as solid-phase antigen and Eu3+-labeled mouse antihuman IgA as corresponding probe. The precision, sensitivity, specificity test, and stability of the TRFIA kit were evaluated, and comparison with the traditional enzyme-linked immunosorbent assay (ELISA) was also investigated. The cutoff value for the TRFIA was 2.5. Intra- and interassay coefficients of variation for the TRFIA were 2.45–3.30% and 3.38–4.61% respectively. There was no cross-reactivity with the antibodies of cytomegalovirus (CMV) or herpes simplex virus (HSV) types 1 and 2, or other potential interferences. The established assay kit also behaved better in sensitivity and stability than the ELISA one. Additionally, the results in 382 serum samples using two analytical methods showed there was good agreement between the TRFIA and commercial ELISA kit. In the current study, the results demonstrated that the TRFIA that was developed for Zta IgA detection was more sensitive and reliable for the diagnosis of EBV infection and had potential value in automation and high-throughput screening.
Introduction
E
There are different typical patterns of antibody response against the various EBV-determined antigens. For example, the typical anti-EBV serological profile of NPC patients consists of an increase in both IgG and IgA antibodies against the viral capsid antigen (VCA), diffuse early antigen (EA-D), and EBV nuclear antigen (EBNA). The typical anti-EBV serological profile of BL patients consists of an increase in IgG against VCA and the restricted early antigen (EA-R) (1). Moreover, the typical anti-EBV serological profile of IM patients consists of an increase in IgM and IgA against VCA, EA-D, and EA-R (11).
ZEBRA, a 40 kDa nuclear protein, is an immediate–early protein that plays an important role in the switch from EBV latency to replication (3,4). Because it expresses quite early in the EBV cycle, the specific antibody against ZEBRA is an important measurable parameter. Fifty percent of Hodgkin's disease, 85% of IM patients, 75–87% of NPC patients, and 32% of human immunodeficiency virus (HIV)-infected patients have ZEBRA antibodies, but in healthy EBV-seropositive donors, it is only 2–4% (6,13,14,18). As described above, the ZEBRA antibody is a sensitive and specific serological marker for detecting EBV-associated disease because it is rare among normal samples (6,27). High levels of anti-ZEBRA antibody could be used not only as a biological marker in the follow-up of NPC and non-Hodgkin's lymphoma patients but also for the prognosis of EBV infectious diseases (22 –24). It has been reported that IgA antibodies could more effectively indicate the risk of NPC (2). Recently, enzyme-linked immunosorbent assay (ELISA) specific for Zta IgA was most commonly used in the clinical diagnosis of EBV infection. However, it offered low sensitivity, and the enzyme-labeled antibodies were unstable.
In this study, the aim was to develop a new immunoassay for Zta IgA detection based on time-resolved fluoroimmunoassay (TRFIA). Compared with the traditional ELISA, the new established assay demonstrated a higher diagnostic sensitivity, more excellent precision, and better stability.
Materials and Methods
Reagents and instrumentation
The Zta antigen was purchased from Innovax (Xiamen, China). Monoclonal mouse antihuman IgA was obtained from the Wuhan Institute of Virology (Chinese Academy of Sciences, Wuhan, China). An Eu3+-labeled kit and enhancement solution were purchased from PerkinElmer Wallac (Turku, Finland). Bovine serum albumin (BSA) was obtained from Bovogen Biologicals Pty Ltd (Melbourne, Australia). Ninety-six-well polystyrene microtiter plates were purchased from Thermo Labsystems (Milford, MA). The commercial ELISA kits for Zta-IgA detection were purchased from Zhongshan Bio-tech Co. Ltd (Zhongshan, China). A Victor™ 1420 time-resolved plate fluorometer was used to analyze TRFIAs (PerkinElmer Life and Analytical Sciences, Waltham, MA). All other chemicals and reagents used were of analytical grade.
Buffers
Coating buffer contained 50 mM/L Tris (pH 7.4) and 0.9% NaCl (v/v). Blocking buffer (pH 7.4) contained 50 mM/L phosphate buffered saline, 0.9% (w/v) NaCl, 0.1% (w/v) BSA, and 0.2% (v/v) Tween-20. Labeling buffer contained 100 mmol/L Na2CO3 at pH 9.0. Elution buffer (pH 7.4) comprised 50 mM Tris-Cl, 0.9% (w/v) NaCl, and 0.2% (w/v) BSA. Assay buffer A (pH 7.8) was made up of 50 mM Tris-Cl, 0.9% (w/v) NaCl, 1.5% (w/v) BSA, and 0.4% (w/v) casein. Assay buffer B (pH 7.8) contained 50 mM Tris-Cl, 0.9% (w/v) NaCl, 0.02% (w/v) BSA, 0.01% (v/v) Tween-20, and 0.05% ProClin 300. The washing buffer (pH 7.8) contained 25 mM Tris-Cl (pH 7.8), 0.9% (w/v) NaCl, and 0.06% (v/v) Tween-20.
Source of human serum specimens
All serum specimens were collected from Guangzhou Daan clinical laboratory center. They were kept at −20°C for storage and further analysis.
Solid-phase antigen preparation
Transparent 96-well plates were coated with Zta antigens (100 μL/well), with a concentration of 2 μg/mL. After incubation overnight at 4°C, the coating buffer was discarded, and the wells were washed three times with washing buffer. Each well was blocked with 200 μL blocking buffer and then incubated at room temperature for 2 h with shaking. After removing the blocking buffer, the plates were dried in a vacuum at 4°C, and stored at 4°C or −20°C in a sealed plastic bag with desiccant for long-term storage.
Antibody labeling
Prior to labeling, 0.5 mg mouse antihuman IgA was purified and concentrated to a final concentration of 2.5 mg/mL. It was gently mixed with 0.1 mg DTTA-Eu (N1-[P-isothiocyannato-benzyl]-diethylene-triamine-N1, N2, N3-tetraacetate-Eu3+) in 200 μL of labeling buffer. Then the mixture was incubated at room temperature overnight with shaking. Labeled mouse antihuman IgA was separated from excess free chelates on a Sephadex G-50 column (1.5 cm×40 cm) using elution buffer. Fractions were monitored at 280 nm to determine concentration. Finally, highly purified BSA was included as a protein stabilizer, and the optimal concentration of the BSA was 1‰. The Eu3+-labeled antibodies were stored at −20°C until used.
Assay protocol
A typical indirect immunoreaction in a two-step assay procedure was adopted. The fundamental of the assay is shown in Figure 1. Serum was diluted with assay buffer A at 1:20, which was added to wells (100 μL/well) coated with Zta antigen, as well as positive and negative controls simultaneously. The strips were incubated with shaking for 45 min at room temperature, and were then washed four times with washing buffer. With an optimal dilution of 1:1,000, 100 μL of Eu3+-labeled mouse antihuman IgA antibody diluted with assay buffer B was added to the wells and incubated with shaking again for 45 min at room temperature. After washing six times, 100 μL enhancement solution was added to each well. After incubation for 5 min, the time-resolved fluorescence intensity of each well was measured by a Victor3 1420 Multilabel Counter.
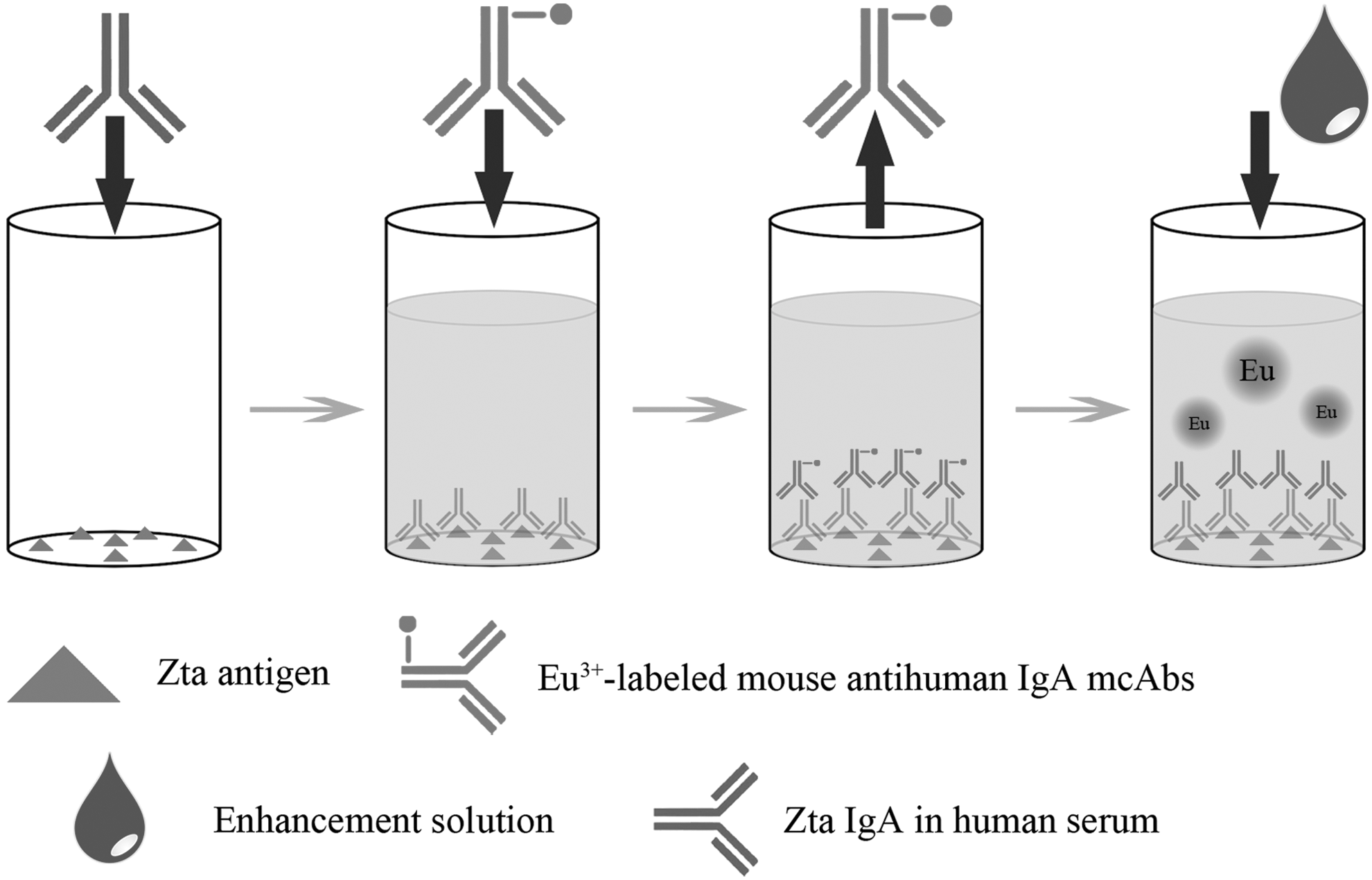
Schematic of the time-resolved fluoroimmunoassay (TRFIA) developed in this study.
Statistical analyses
Origin 75 software was used for statistical analysis. Sample means and standard deviations were obtained using Microsoft Excel. The coefficient of variation was employed to analyze the intra- and interassay variation.
Results
Optimization of indirect TRFIA
In this study, a new indirect TRFIA was developed. As is well known, there were some reactive parameters, including coating concentration, serum dilution, and the dilution ratio of Eu3+-labeled antibody, which significantly influenced the analytical performance.
The optimal condition of coating concentration of the antigen and the dilution ratio of Eu3+-labeled antibody were determined by cross-titration. The antigens and Eu3+-labeled antibodies were diluted from 4 μg/mL to 0.5 μg/mL and 1:400 to 1:6,000 respectively. As shown in Figure 2, the fluorescence values of the positive and negative samples tended to be stable when the coating concentration reached 2 μg/mL, and it could distinguish the positive and negative samples clearly when the dilution of Eu3+-labeled antibody was 1:1,000 (Fig. 3). The optimal serum dilution was investigated using several serum samples diluted from 1:10 to 1:2,560 with assay buffer A. The result showed that the intensity of fluorescence decreased as the serum dilution ratio increased. Finally, the optimal coating concentration was found to be 2 μg/mL, the optimal dilution of serum was 1:20, and that of Eu3+ labeled antibody was 1:1,000.
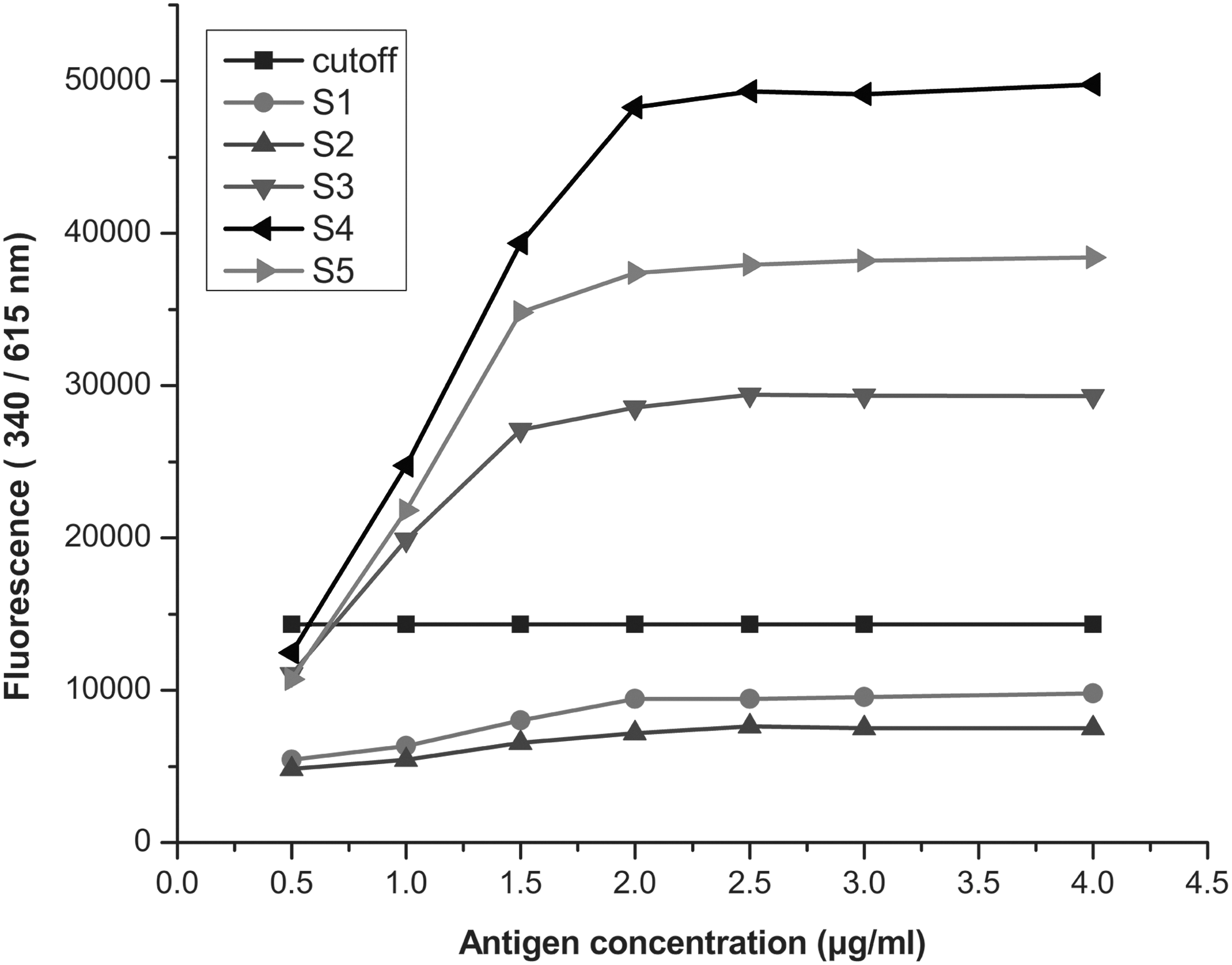
Influence of coating concentration on fluorescence intensity. The Zta antigen was diluted from 4.0 to 0.5 μg/mL. Samples S1 and S2 were negative, while S3, S4, and S5 were positive.
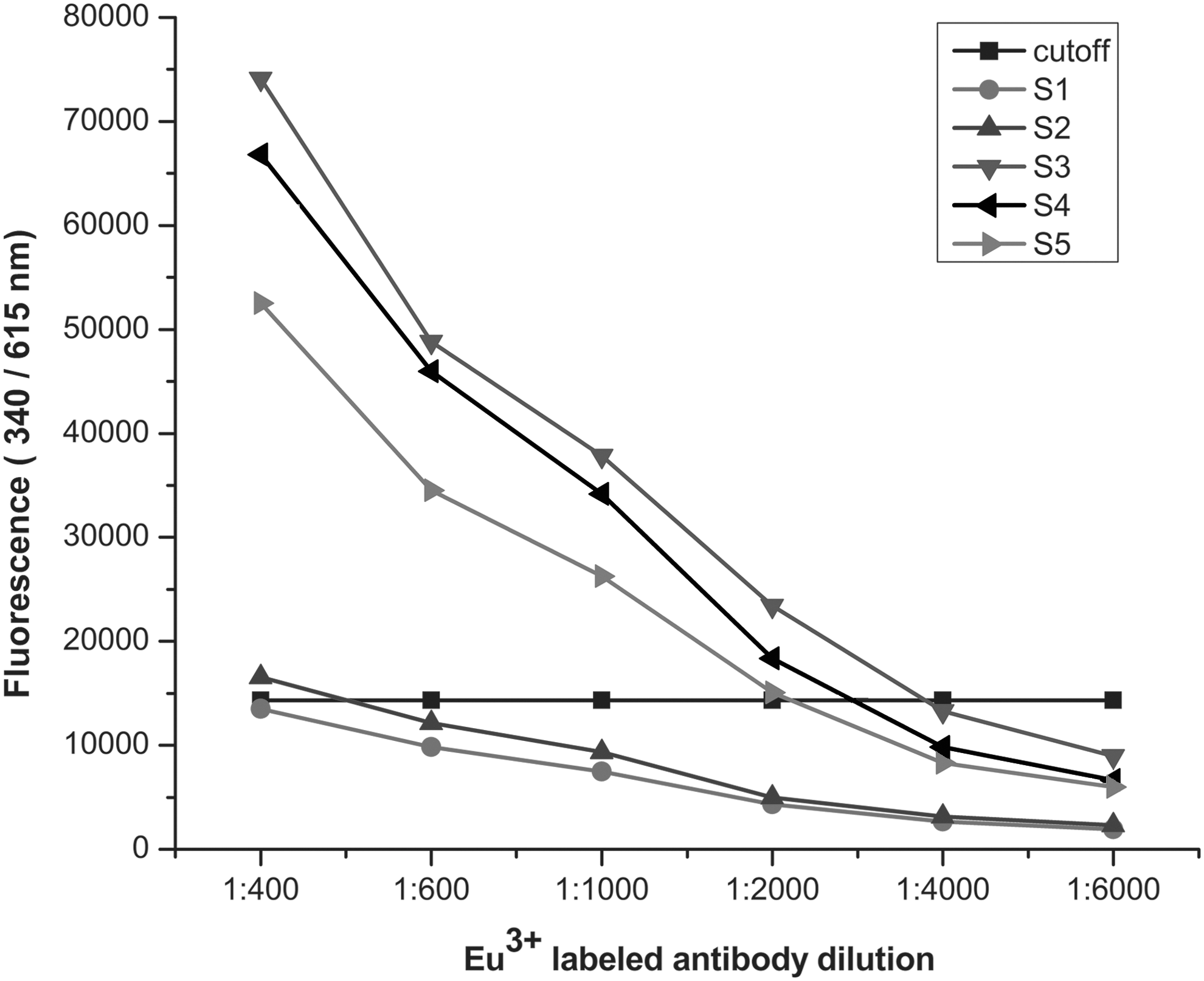
Influence of Eu3+-labeled antibody dilution on fluorescence intensity. Eu3+-labeled mouse antihuman IgA was diluted 1:400–1:6,000. Samples S1 and S2 were negative, while S3, S4, and S5 were positive.
Cutoff value
A total of 370 healthy samples were tested. The cutoff value of the TRFIA kit was 2.5, which was the ratio between fluorescence of sample and that of the negative control. The ratio ranged from 0 to 2.99, and in 99.2% of samples the ratio was <2.5. Therefore, a sample was considered negative when the ratio was <2.5. Otherwise, the sample was considered positive.
Precision
Intra- and interassay coefficients of variation (CVs) were conducted by measuring three human serum samples (S1, S2, and S3). As shown in Tables 1 and 2, the intra- and interassay CVs detected by TRFIA were 2.45–3.3% (n=10) and 3.38–4.61% (n=12) respectively, whereas those for ELISA were 3.64–8.82% (n=10) and 6.40–11.63% (n=12).
TRFIA, time-resolved fluoroimmunoassay; ELISA, enzyme-linked immunosorbent assay; SD, standard deviation; CV, coefficients of variation.
Sensitivity
The sensitivity of the TRFIA was evaluated using a positive sample diluted from 1:20 to 1:2,560 with assay buffer A. The samples at different dilution ratios were tested in duplicate, and the positive and negative controls were simultaneously measured. The results were compared and analyzed with ELISA (Table 3). The sample was considered positive when diluted 1:20–1:320, and negative at the dilution ratio of 1:640 by TRFIA. In comparison, the sample was considered positive when diluted 1:20–1:80, and negative when diluted 1:160–1:640 by ELISA.
Specificity test
The specificity of the TRFIA was evaluated by detection of the antibodies of potential interferences, namely, cytomegalovirus (CMV), HSV-I and HSV-II, human papilloma virus (HPV), hepatitis virus (HAV), hepatitis B virus (HBV), hepatitis C virus (HCV), and HIV. The results demonstrated that there were negligible effects on the concurrent assay for Zta IgA; even the interfering compounds were at high levels.
Stability of the TRFIA kits
The TRFIA kit was put into the 37°C constant temperature box for 7 days. The positive and negative controls were tested, and it was found that there were no obvious changes of fluorescence compared with those stored at 4°C. This result demonstrated that the TRFIA kit satisfied the stability requirement and could be stored at 4°C for 1 year.
Comparison of TRFIA and ELISA
In order to investigate the reliability of the established TRFIA immunoassay for clinical application, a total of 382 clinical serum samples were analyzed. The dates were compared with those obtained by ELISA. As shown in Table 4, the sensitivity and specificity of the TRFIA kit were 98.8% and 98.2%, respectively.
Discussion
EBV, the best-known member of the gamma-1 herpesviruses, infects >90% of the adult population worldwide (15,21). Due to the susceptibility of humans to EBV and the close association with a variety of human disease, especially malignant tumors, the detection of EBV infection has been given increasing attention in recent years. Clinically, serological tests for EBV have been used as diagnostic predictors of EBV-associated diseases for many years (19,26). EBV antibodies are measured by several methods, including immunoblot strip, immunofluorescence assay (IFA), and ELISA. Immunoblot strip is used as a confirmation test. The preparation and analysis of the approach are laborious and cumbersome, which limits its application in large-scale screening (7). IFA covers IgM-VCA, IgG-VCA, IgG-EA-D, IgG-EA-R, and anti-EBNA detection. Compared to other techniques, it is the most sensitive and specific assay and is still used as a “gold standard.” However, it also has some disadvantages. The lymphoblastoid cells, as the source of the diagnostic antigen, are expensive and difficult to maintain. Moreover, the result is subjective because the test mostly relies on a skilled technician. In addition, it is not suitable for mass screening (10). ELISA, a method commonly used in clinical conditions, is characterized by objective measurement, standardization, and automatization. However, it has a low sensitivity, and the enzyme-labeled antibodies used in the assay could become unstable (5,10). Therefore, in order to diagnose EBV-associated diseases more effectively and accurately, it is necessary to develop a new molecular diagnostic technique.
Herein, a TRFIA was established to detect the ZEBRA IgA. TRFIA, a novel technology that has rapidly developed in recent years, is characteristic of wide detection range, high sensitivity, long fluorescence lifetime, substantial Stokes shift, easy automation, less susceptibility to matrix interference, and no radioisotope pollution (12,16). It offers an almost ideal way for the determination of many biomarkers. In this study, compared with a commercial ELISA, the performance characteristics of TRFIA were equivalent or better in many respects. In the sensitivity test, the results indicated that the Zta IgA TRFIA that was developed had a wider detectable range than the commercial ELISA. The results of the precision and cross-reactivity tests demonstrated that the TRFIA was reliable and specific for the determination of Zta IgA in human serum. Additionally, due to the independence of the disintegration of the labeling compound, the TRFIA had an obvious superiority in stability. In the TRFIA kit, after adding an enhanced solution to the wells, the fluorescent intensity varied little, even though the microplates were installed overnight. However, in the commercial ELISA kit, after adding the stop solution, the optical densities (ODs) decreased with time and could hardly last through 30 minutes.
In conclusion, first, an indirect TRFIA was developed for the detection of Zta IgA antibodies in human serum. This assay has high precision, excellent sensitivity, and good repeatability. There was good agreement between the TRFIA and commercial ELISA, which indicated the TRFIA kit has good clinical applicability. Furthermore, due to the suitability for use in the PE AUTODELFIA™ 1235 automatic system, the assay has potential value to automation and high-throughput screening.
Footnotes
Acknowledgments
This work was supported by Guangzhou major blood-borne infectious disease molecular diagnostic reagents R&D and industrialization (Grant No. 2014Y2-00002).
Author Disclosure Statement
No competing financial interests exist.