
Abstract
Multispecific, broad, and potent T cell responses have been correlated with viral clearance in hepatitis C virus (HCV) infection. However, the majority of infected patients develop chronic infection, suggesting that natural infection mostly leads to development of inefficient T cell immunity. Multiple mechanisms of immune modulation and evasion have been shown in HCV infection through various investigations. This study examined the generation and modulation of T cell responses against core and frameshift (F) proteins of HCV. A single immunization of mice with replication incompetent recombinant adenovirus vectors encoding for F or core antigens induces poor T cell responses and leads to generation of CD4+ and CD8+ T cells with low granzyme B (GrB) expression. These T cells have impaired GrB enzyme activity and are unable to kill peptide loaded target cells. The low intracellular expression of GrB is not due to degranulation of cytotoxic granules containing cytotoxic T cells. Addition of exogenous IL-2 in in vitro cultures leads to partial recovery of GrB production, whereas immunization with the Toll-like receptor (TLR) agonist poly I:C leads to complete restoration of GrB expression in both CD4+ and CD8+ T cells. Thus, a possible new strategy of T cell modulation is recognized wherein effector T cells are caused to be dysfunctional by HCV-derived antigens F or core, and strategies are also delineated to overcome this dysfunction. These studies are important in the investigation of prophylactic vaccine and immunotherapy strategies for HCV infection.
Introduction
P
HCV is a single-stranded positive-sense RNA virus belonging to the Flaviviridae family and the Hepacivirus genus (64). It has seven major genotypes, and each genotype has several subtypes (29). Within an individual host, large numbers of quasispecies of HCV exist. Error-prone RNA-dependent RNA polymerase causes the emergence of escape mutants resistant to neutralizing antibody and CD8+ cytolytic T lymphocytes (22,27). Design of a preventive or therapeutic vaccine is a major challenge for HCV due to the high genetic variability of the virus and a lack of understanding of protective immune responses. However, there is encouraging evidence of protective immunity in HCV pathogenesis: 20–35% of infected people spontaneously clear the virus due to induction of efficient host immune responses, which suggests that the design of a vaccine capable of clearing the virus from the host is possible (53). In chimpanzee studies, it has been shown that both antibody and cellular immune responses are partially effective in controlling HCV infection (13,24,30). However, chronic/persistent infection in the majority (65–80%) of infected people suggests that this virus has devised multiple strategies to evade the immune system and induced immunity. The factors and mechanisms that allow the virus to circumvent the host's immune responses and to persist in infected individuals are not clearly understood. Various mechanisms of T cell failure have been suggested, such as impaired primary T cell activation, T cell exhaustion, T cell dysfunction, impaired T cell maturation, suppression of T cell function by viral factors, unresponsiveness due to exposure to high antigen levels, impaired dendritic cell (DC) functions, and suppression of T cell function by regulatory T cells (9,19,26,32,41,44,55).
The HCV genome encodes a single polyprotein precursor of ∼3010 aa (12), which, after processing by host and viral proteases, generates 10 different viral proteins: the core, E1, E2, p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B (80). In addition, a protein within the core encoded region, the frameshift (F) or alternate reading frame protein (ARFP), has been reported to be expressed from an alternative +1 reading frame (75,77,80). The F protein is localized in the endoplasmic reticulum (ER) region, implying that it has a role in viral morphogenesis (81). Previous work suggests that the F protein is expressed in natural HCV infections (16). B and T cell mediated immune responses specific to ARFP have been detected in HCV patients (4). It has also been reported that anti-F antibody is significantly increased in patients with HCC, suggesting a possible role and/or presence of F protein during carcinogenesis (16). The F protein is not essential for viral replication in cell culture or in vivo (56), but its role in disease progression and in the development of HCC has not been ruled out.
The core protein of HCV has been implicated to have an immune modulatory role in several studies. The infection of mice with recombinant vaccinia virus expressing HCV core was shown to suppress host immune responses by inhibiting antiviral cytotoxic T lymphocyte activity, and production of IL-2 and IFN-γ (46). Recombinant core protein has recently been shown to suppress type I and III IFN production in plasmacytoid DCs in response to TLR agonists, which was linked to decreased IRF7 protein levels and increased non-phosphorylated STAT1 protein (71). It has previously been shown that recombinant core protein and polyprotein of HCV inhibit NF-kB activation in human DCs (44). Further, transfection of Huh-7 cells with a plasmid encoding the HCV core protein attenuated TNF-α induced NF-kB activation and enhanced TNF-α induced cell death (61). Expression of core protein in a human CD4+ JURKAT T cell line led to upregulation of FoxP3 and CTLA-4, and an ability to suppress CD4+ and CD8+ T cell responses (18). Ligation of gC1qR onto human monocyte-derived DCs with HCV core has been shown to inhibit TLR-induced IL-12 production and cause a reduced ability to stimulate allogeneic CD4+ T cells (76). Core protein has also been shown to suppress interferon regulatory factor-1 (IRF-1) transcription and to repress several IFN-stimulated genes such as those encoding IL-15 and IL-12 (14). Expression of core protein in a human hepatocyte cell line HepG2 led to regulation of NF-kB activation, suppression of apoptosis, and the inflammatory response (60). In addition, core protein has been shown to suppress IL-12 synthesis selectively in human macrophages by interfering with AP-1 activation (20). Therefore, in multiple studies, core protein of HCV has been shown to regulate/suppress innate and adaptive immune responses by various mechanisms.
In the authors' earlier studies, it was shown that HCV-derived antigens NS3 and core play disparate roles in the development of effector T cells and in the reduction of viral titers in a recombinant-HCV-vaccinia infection model (45). Immunization with recombinant adenovirus vector containing HCV core led to suppression of the host immune response by developing Tregs, reducing proliferation of T cells, inducing lower numbers of GrB+ effector cells, and producing lower levels of effector cytokines and high levels of IL-10. In contrast, efficient effector T cell responses were generated in response to immunization with recombinant adenovirus vector containing HCV-NS3 in the same infection model (45). These differences in induced immune responses against HCV core and NS3 antigens translated to distinct outcomes in viral infection in an HCV-vaccinia surrogate infection model: immunization with NS3 led to reduced viral titers in contrast to immunization with core (45).
The current study examined T cell responses generated in mice against F or core antigens using individual recombinant adenovirus vectors encoding for each of these proteins. The results demonstrate that T cells obtained from mice immunized with either F or core proliferate weakly against the respective antigen-derived peptides. Interestingly, high levels of IL-10 and low levels of IFN-γ were produced in peptide-stimulated cultures. It was observed that expression of intracellular granzyme B (GrB) in CD4+ and CD8+ T cells obtained from mice immunized with F or core was much lower than expression in a control vector (CV) or in NS3 immunized mice. Diminished expression of GrB by CD8+ T cells from mice immunized with F or core correlated with reduced GrB enzyme activity and with reduced killing of peptide-loaded EL-4 target cells. It was observed that immunization of mice with adenovirus vectors containing F or core, together with a Toll-like receptor-3 (TLR-3) ligand poly I:C, resulted in recovered GrB expression levels in T cells, and also caused splenocytes cultured with exogenous IL-2 in vitro to demonstrate increased GrB expression in both CD4+ and CD8+ T cells. These studies reveal downregulation of GrB expression as a possible means of modulating effector T cell responses in a chronic infectious disease. This might represent a new strategy of immune evasion by HCV.
Materials and Methods
Mice
Male C57bl/6 mice, 6–8 weeks old, were purchased from Charles River Laboratories, Inc. (Canada). All animal experimental protocols were approved by the University of Alberta Animal Care and Use Committee for Health and Sciences, and conducted in accordance with Canadian Council of Animal Care (CCAC), Canada, guidelines.
Recombinant adenovirus vectors containing NS3, core, and a no antigen CV have been described by the authors (45,51). The F protein was polymerase chain reaction (PCR) amplified from full-length clones of HCV H77 cDNA using forward primer 5′-GAA GAT CTA TGC CAA ACG TAA CAC CAA CCG TC-3′ and reverse primer 5′- GAA GAT CTC ACG CCG TCT TCC AGA ACC CGG A-3′ and used to prepare a recombinant adenovirus vector using methodologies described by the authors (51). The adenovirus vectors were propagated and amplified in 293A cells, and the presence of the F gene was confirmed by sequencing.
Immunization of mice
Mice were injected intramuscularly (i.m.) in the quadriceps muscles of both hind limbs with 100 μL of 2×107/mouse replication-deficient recombinant adenovirus particles expressing F, core, NS3, or CV. (Hereafter can be denoted simply as F, Core, NS3 or CV immunization or immunized mice.) For single immunizations, mice were injected on day 0. For three immunizations, mice were injected on days 0, 10, and 20. The NS3 immunization group was used as a positive control in most of the experiments. In co-immunization experiments, both F or core and NS3 were injected i.m. in both hind limbs with a total of 2×107/mouse replication-deficient recombinant adenovirus particles. Immunization with poly I:C (polyinosinic: polycytidylic acid, Sigma-Aldrich, St. Louis, MO) used at 20 μg/mouse mixed in phosphate buffered saline (PBS) in 100 μL.
Isolation of splenic T cells
On day 8 after the last immunization, mice were euthanized to allow harvesting of splenocytes. The spleens of mice from each group were pooled into one suspension of cells, which was prepared by disrupting each spleen between frosted slides and filtering the contents through a Falcon 100 μm nylon cell strainer. After centrifugation, red blood cells were lysed, and T cells were enriched using nylon wool columns as described previously (45). These enriched T cells (∼90% CD3+ T cells) were used in various experiments.
T cell proliferation assay
Proliferative responses of splenic T cells were measured in triplicate cultures in 96-well flat-bottomed microtiter plates. A total of 4×105 T cells from F- or core immunized mice and 4×105 antigen presenting cells (APCs; spleen cells from naïve unimmunized mice irradiated with 18 Gy) were mixed with 1 μg/mL and 10 μg/mL synthetic peptides (15-mers; Tables 1 –3) derived from F, core, or NS3 proteins (GenScript, Piscataway, NJ) and cultured in complete RPMI medium (Hyclone Laboratories, South Logan, UT) containing 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA) at 37°C and in 5% CO2 for 4 days. Media and Con A (1 μg/mL) were used as negative and positive controls, respectively. The wells were pulsed with 0.5 μCi/well [3H]-thymidine (GE Healthcare, Morgan Boulevard, Canada) for 12–18 h and harvested on filter papers (PerkinElmer, Waltham, MA) using a 96-well harvester (Tomtech, Hamden, CT). Measurement of [3H]-thymidine incorporated into the DNA of proliferating cells was performed in a Microbeta Trilux liquid scintillation counter (PerkinElmer). Stimulation indices were calculated by dividing the counts per minute (CPM) obtained in the presence of peptides with those in the dimethyl sulfoxide (DMSO) control (solvent for dissolving peptides). Proliferation is represented as the mean stimulation index±standard error (SE) of triplicate cultures from a representative experiment of three to six repeated experiments.
HCV, hepatitis C virus.
Cytokine enzyme-linked immunosorbent assay
Cytokines IL-10, IFN-γ, TNF-α, TGF-β, and IL-12 secreted into the supernatant of proliferating cultures were measured using sandwich enzyme-linked immunosorbent assay (ELISA) kits following the manufacturer's protocol (eBioscience, San Diego, CA). Dilutions of 1:5 to 1:20 were used for the samples, with standards ranging from 15.6 to 2,000 pg/mL. ELISA plates were read, and the concentrations of cytokines were calculated with an automated ELISA plate reader (Fluostar Optima; BMG Labtech, Allmendgruen, Germany).
Flow cytometry analysis of intracellular cytokines, GrB, and surface markers
A total of 1×106 splenocytes/mL from mice immunized with F, core, NS3, or CV were cultured in vitro for 5 days in complete RPMI media with mixtures of peptides from respective proteins (F [1–16], core [1–16], or NS3 [1–8], each at 1 μg/mL concentration; Tables 1 –3). Controls had no added peptide (CV group). On day 5, cells were collected for extracellular and intracellular staining with fluorescently labeled mAbs (concentrations according to manufacturer's instructions). Briefly, the cells were washed with ice-cold FACS buffer (PBS with 2% FBS) and antimouse antibodies were added for extracellular staining (CD3e-PE, CD4-PE-Cy5, CD8aAPC-eFluor 780, and CD107b-FITC; eBioscience). The cells were incubated at 4°C for 30 min. The cells were washed twice with FACS buffer and fixed in 2% paraformaldehyde for 10 min. For intracellular staining, the cells were incubated in cold FACS buffer+ 0.3% saponin (Sigma-Aldrich) for 5 min, followed by addition of anti-GrB-Alexa fluor 647 (Biolegend, San Diego, CA) and incubated for 45 min at 4°C. The cells were washed twice with 0.3% saponin containing FACS buffer followed by washing twice with FACS buffer. The cells were also stained with isotype control antibodies using similar methodologies, run in a FACS-Canto, and analyzed using FACS-DIVA software (Becton Dickinson, Mountain View, CA). The cells were gated to exclude 98% of isotype-matched control monoclonal antibody-stained cells. For the analysis, lymphocytes were gated based on side scatter (SSC) and forward scatter (FSC), followed by gating for CD3+ T cells. The CD3+ T cells were then gated on either CD4+ or CD8+ T cells, followed by examining GrB expression, which is shown as histograms in most of the figures.
Intracellular staining for IL-10
A total of 1×106 splenocytes/mL from F, core, NS3, or CV immunized mice were cultured in vitro for 5 days in complete RPMI media with mixtures of peptides from respective proteins or no peptides (for CV group) as described above. These splenocytes were restimulated with PMA and ionomycin (Sigma-Aldrich) at 5 ng/mL and 500 ng/mL, respectively, for 4 h in the presence of Brefeldin A (3 μg/mL; eBioscience). After incubation, cells were stained extracellularly with antimouse CD3-PE-Cy7, CD4-PE-Cy5, CD8aAPC-eFluor 780, and intracellularly with anti-IL-10-FITC (eBioscience), using the procedure described in the previous section. The control cells were stained with isotype matched antibodies. The cells were run on FACS-Canto, and analyzed using FACS-DIVA software (Becton Dickinson). The cells were gated to exclude 98% of isotype-matched control antibody staining and analyzed for each marker. To analyze the phenotype of cells, lymphocytes were gated based on SSC and FSC, followed by gating for CD3+ T cells. The CD3+ T cells were then gated on either CD4+ or CD8+ T cells, followed by examining IL-10 expression, which is shown as histograms in the figure (S1).
GrB enzyme assay
GrB was assayed according to methods reported previously (67), briefly, from a 5-day in vitro culture of splenocytes obtained from mice immunized with various recombinant adenovirus vectors with 1 μg/mL of respective peptide mixture or no peptide for the CV group. CD4+ and CD8+ T cells were purified from the 5-day splenocyte cultures using antibodies (EasySep, Mouse CD4+ and CD8+ T cell selection kit; StemCell Technologies, Vancouver, BC) according to the manufacturer's instructions. A total of 2×106 viable splenocytes, CD4+, or CD8+ T cells were resuspended in 50 μL of lysis buffer (150 mM NaCl, 20 mM Tris, pH 7.2, 1% [v/v] Triton X-100) for 10 min on ice. Supernatants were collected following a 10-min micro-centrifugation (10,000 g). Fifty μL of each lysate was preincubated with the pancaspase inhibitor Z-VAD-FMK (Enzo Life Sciences, Farmingdale, NY) for 30 min before addition of the GrB substrate. The paranitroanilide substrate, acetyl-Ile-Glu-Thr-Asp-paranitroanilide (Ac-IETD-pNA; Enzo Life Sciences) was used at 200 μM in reaction buffer containing 50 mM HEPES (pH 7.5), 10% (w/v) sucrose, 0.05% (w/v) CHAPS, and 5 mM DTT. GrB activity was determined by hydrolysis of the substrate at 37°C in 96-well flat-bottom tissue culture plates (Nalgene Nunc International, Penfield, NY) in a final volume of 100 μL. Released paranitroanilides were measured with a spectrophotometer (Fluostar Optima, BMG Labtech) at 405 nm in a time-course manner. Enzymatic activity was quantified using a standard curve of recombinant mouse GrB (Enzo Life SciencesNY) activity with dilutions of 2–200 IU/mL and normalized to total protein content.
CFSE-based EL-4 killing assay
The cytolytic activity of the splenocytes obtained from immunized mice, and then cultured with peptides as described in the flow cytometry section above, was measured using EL-4 as target cells at a 30:1 effector-to-target ratio in a 4 h CFSE loss assay. EL-4 cells were propagated for two or three passages and incubated overnight with pools of synthetic peptides (1 μg/mL each) derived from F, core, or NS3 proteins separately. For splenocytes from CV immunized mice, EL-4 target cells were loaded with mixtures of peptides from F, core, and NS3 proteins. EL-4 cells were harvested and stained with 2 mM of CFSE (5- and 6-carboxyfluorescein diacetate succinimidyl ester or CFDA SE; Invitrogen) in PBS for 15 min in the dark. CFSE-stained target EL-4 cells were mixed with effector splenocytes in a 1:30 ratio and incubated for 4 h at 37°C in an atmosphere of 5% CO2. After 4 h, 20,000 Count Bright beads (Invitrogen) were added to each tube. Using FACS Diva to gate the beads, the flow cytometer was run to collect 10,000 beads from each tube to standardize the amount of cells tested in each tube. The difference in CFSE-stained cells between treatment and control tubes indicate the number of cells that were killed. Data are represented as numbers of dead cells in each culture (normalized by a constant number of beads).
Reversal of GrB expression by IL-2 treatment in vitro
Splenocytes from immunized mice were cultured with mixtures of respective peptides (1 μg/mL each) in the presence or absence of murine recombinant IL-2 at 100 pg/mL (Biosource, Grand Island, NY) for 5 days. Cells were then stained as described before with CD3e-FITC, CD4-PE-Cy5, CD8-APC-eFluor 780, and GrB-Alexa Fluor 780 (eBioscience), followed by flow cytometry analyses of stained cells.
Suppression of GrB expression by inhibiting NF-κB activity
Splenocytes obtained from mice immunized with various recombinant adenovirus vectors were cultured with respective peptides at 1 μg/mL in the presence or absence of a chemical inhibitor of NF-κB, pyrrolidine dithiocarbamate (PDTC), at 10 μM for 5 days, followed by staining with fluorescently labeled antibodies and flow cytometry analyses.
Statistical analysis
Statistical analysis was done using one-way analysis of variance using GraphPad Prism (GraphPad Software, Inc., La Jolla, CA). A single asterisk indicates a significant difference at p<0.05, and a triple asterisk indicates a significant difference at p<0.0007.
Results
T cells from mice immunized with recombinant adenovirus vectors expressing F or core antigens of HCV do not show peptide-specific proliferation
To study the generation of immune responses against HCV-derived F and core antigens, C57Bl/6 mice were immunized with 2×107 recombinant adenovirus particles containing F or core antigens. Mice immunized with recombinant adenovirus vector containing NS3 were used as a positive control. Eight days after a single immunization, splenic T cells were isolated and cultured in vitro with APCs in the presence of 1 and 10 μg/mL of synthetic peptides from respective proteins. The synthetic peptides used in this study are listed in Tables 1 –3. Cells collected from mice immunized with adenovirus vectors containing F or core showed no proliferation, or low levels of proliferation, against all of the peptides tested (Fig. 1). In contrast, cells from NS3 immunized mice exhibited significantly higher proliferation in response to incubation with 1 μg/mL of selected synthetic peptides (Fig. 1) (45). These results suggest that F and core antigens are either poorly immunogenic in mice or that they downregulate immune responses. Proliferative responses to a mitogen such as concanavalin A (Con A) were similar in splenocytes obtained from F, core, NS3, or CV immunized mice (Fig. 1). Interestingly, similar results were obtained after three immunizations. Proliferative responses against F- and core-derived peptides were not substantially increased and were at the same level as those after a single immunization (data not shown). The proliferative response of inguinal lymph node cells from mice immunized with HCV-derived F, core or NS3 proteins was also determined, and similar results were found as for splenocytes (data not shown).
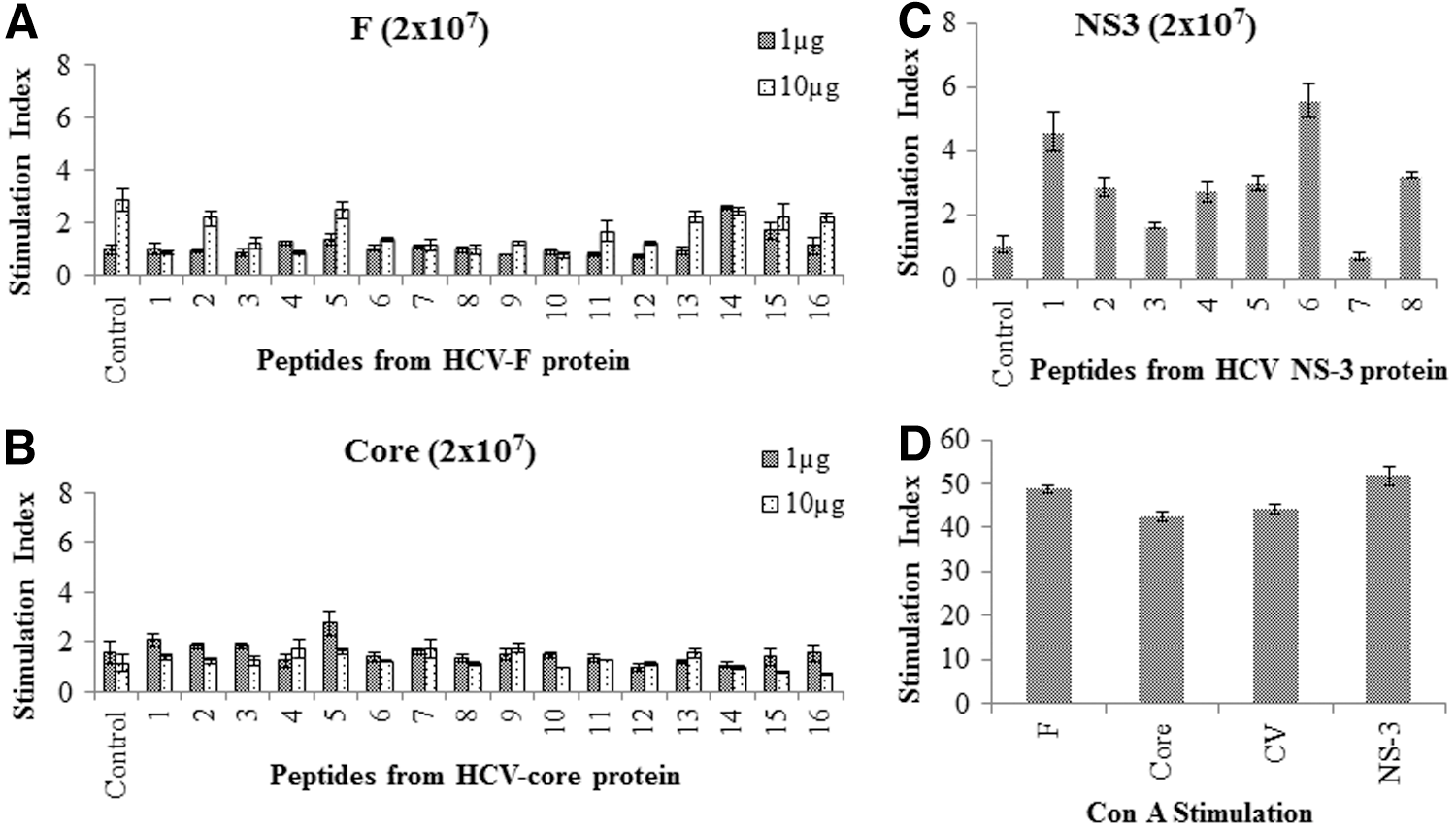
Antigen-specific proliferation of T cells after a single intramuscular (i.m.) immunization with F, core, or NS3. Peptide specific proliferation of T cells after a single i.m. immunization of mice with 2×107 recombinant adenovectors containing F, core, or NS3 antigens. Eight days after, mice were euthanized, and T cells were isolated from spleens and cultured in vitro for 4 days with irradiated syngeneic APCs in the presence of respective overlapping peptides from F or core at 1 and 10 μg/mL, or NS3 peptides at 1 μg/mL. The numbers on the x-axis of the graphs represent the peptide number described in Tables 1
–3. T cells and APCs in the absence of peptides served as controls. Cells cultured in the presence of comparable concentrations of dimethyl sulfoxide (DMSO) were used as background to determine the stimulation indices (stimulation index=counts per minute [CPM] obtained in the presence of peptides/CPM obtained in the presence of solvent DMSO control). (
The production of cytokines IL-10, IFN-γ, TGF-β, IL-12, and TNF-α was examined in the culture supernatants. T cells from mice immunized with F or core, when cultured with respective antigen-derived peptides at both 1 and 10 μg/mL concentrations, produced high levels of IL-10 compared with the control group cultured with no peptide (data from 1 μg/mL of peptides are shown in Fig. 2). No IFN-γ or very low levels of IFN-γ were detected in these cultures (Fig. 2.). In the authors' previous report, it was demonstrated that splenocytes obtained from core immunized mice, when restimulated with recombinant core protein, produced high levels of IL-10 and low levels of IFN-γ (45). Antigen dependent IL-10 expression was also analyzed from CD4+ and CD8+ T cells using intracellular cytokine staining (Supplementary Fig. S1; Supplementary Data are available online at
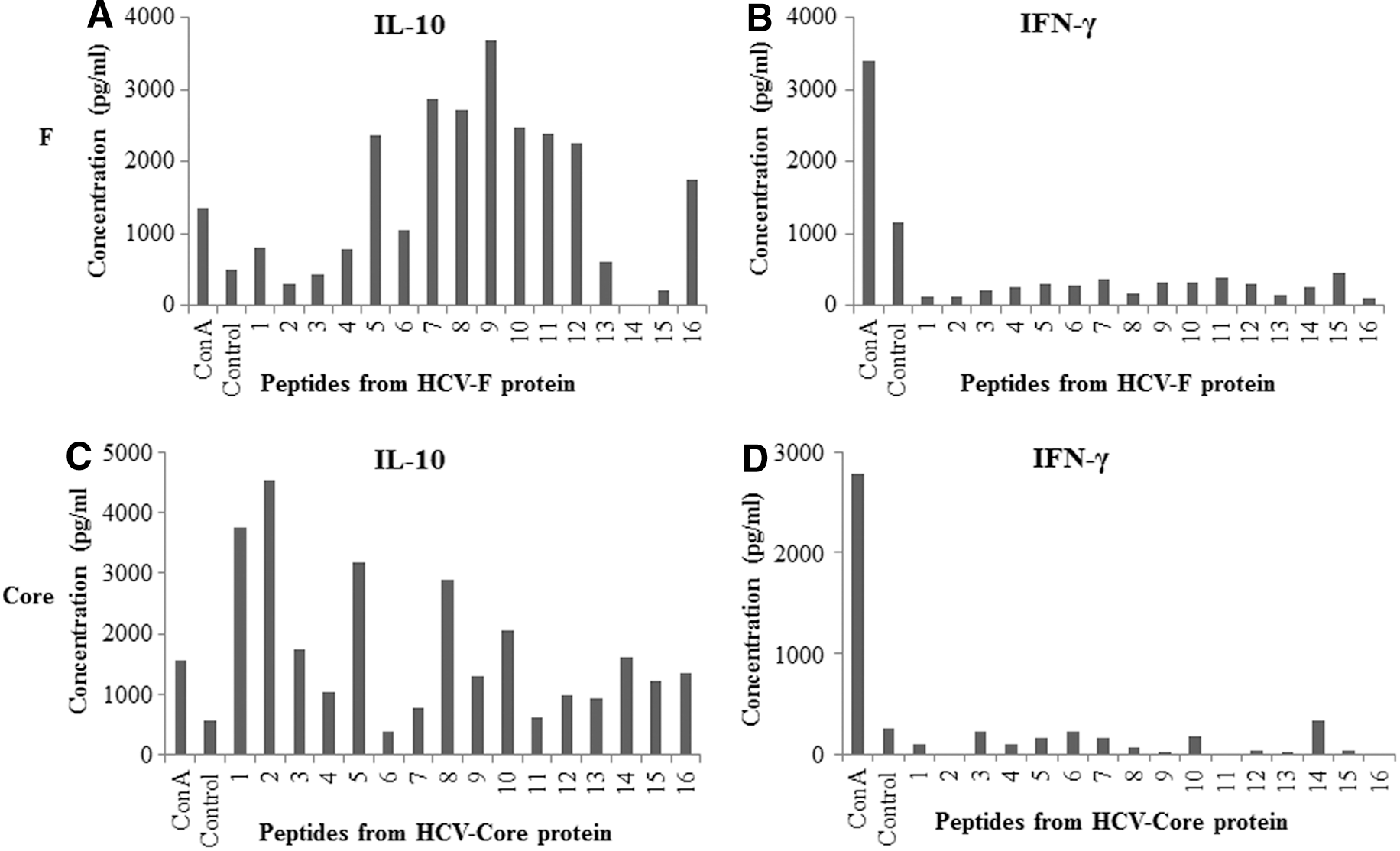
T cell cultures stimulated with F- or core-derived overlapping peptides produce high levels of IL-10 and very low or undetectable IFN-γ. T cells obtained from spleens of mice immunized with 2×107 recombinant adenovectors containing F or core were cultured with irradiated APCs and 1 μg/mL of respective peptides as described in Figure 1. Supernatants from T cell cultures were collected on day 4, and cytokines were analyzed by enzyme-linked immunosorbent assay (ELISA). Assays were done in duplicate, and average values are shown. Cultures without peptides represent negative controls. Cultures with Con A represent the positive control. The numbers on the x-axis of the graphs represent the peptide number as described in Tables 1
–3.
Immunization with F or core antigens of HCV results in reduced GrB expression in CD4+ and CD8+ T cells
To determine the effector function of T cells stimulated after in vivo immunization with F or core antigens, the production/expression of GrB within these cells was examined. Whole splenocytes obtained after one or three immunizations were cultured in vitro for 5 days in complete media in the presence of 1 μg/mL peptide mixture of their respective protein, followed by staining for CD3, CD4, CD8, and intracellular GrB. After 5 days of culture of splenocytes from F, core, NS3, and CV immunized mice, we obtained similar numbers and viability of cells (33–40% yield with 74–83% viability) in different groups and from different experiments with no specific trend. In contrast, splenocytes obtained from PBS sham immunized mice provided a yield of <2% after culture for 5 days in complete media. Percentages of CD4+ GrB+ and CD8+ GrB+ T cells were observed to be significantly lower in mice immunized with either F or core compared with mice immunized with CV (Fig. 3). In contrast, splenocytes from mice immunized with positive control NS3 showed significantly higher expression of GrB in both CD4+ and CD8+ T cells. The up- and downregulation of GrB in T cells obtained from mice immunized with NS3 and core, respectively, is in line with the authors' previous report (45). However, here F protein is also demonstrated to downregulate GrB expression. Notably, the results of one and three immunizations were similar in all of the antigens and CV groups (data not shown). Based on this different role of F or core versus NS3 (in inducing effector T cells with reduced or normal GrB+ T cells), this study sought to determine if co-immunization with NS3+ core or NS3+ F antigens would lead to T cells with reduced GrB expression (Supplementary Fig. S2). In mice co-immunized with F/core and NS3, the induction of CD4+ and CD8+ T cells with reduced GrB was observed. This was similar to the result obtained with T cells collected from mice immunized with F or core alone (Supplementary Fig. S2). This suggests that immune modulation induced by F or core immunization has a significant impact on the induction of potent effector functions of NS3-specific T cells.

GrB production is reduced in mouse T cells after immunization with adenovirus vectors containing recombinant HCV F or core. Splenocytes obtained from immunized mice were cultured for 5 days in vitro in the presence of 1 μg/mL mixtures of peptides (F [1–16], core [1–16], or NS3 [1–8]; Tables 1
–3) from respective proteins or with no added peptide (for CV group). After 5 days, cells were stained for extracellular CD3, CD4, CD8, and intracellular GrB. Lymphocytes were gated based on side scatter (SSC) and forward scatter (FSC). T cells were first gated based on CD3+ expression and then further gated based on CD4+ or CD8+ expression. (
Degranulation of T cells containing granzyme could lead to reduced intracellular levels of GrB in splenocytes. To determine whether reduced intracellular GrB levels in T cells was due to lower expression of GrB or degranulation, the splenocytes obtained from mice immunized with F or core were stained with an antibody to CD107b (LAMP-2) after culture for 5 days in vitro with peptide mixture. CD107b is a cell surface marker that indicates degranulation of cytotoxic T cells. It was found that CD107b expression was similar in CD4+ and CD8+ T cells obtained from F, core, and CV groups, but was slightly higher in cells from the NS3 group (Supplementary Fig. S3). These results confirm that reduction in GrB expression in T cells from mice immunized with F or core is due to differential expression of GrB rather than degranulation of GrB containing granules.
Enzyme activity of GrB is reduced in T cells obtained from F or core immunized mice
To confirm that the expression of GrB in T cells corresponds to GrB enzyme activity, a synthetic substrate Ac-IEPD-pNA was used (21). Cell lysates prepared from splenocytes or purified CD4+ and CD8+ T cells obtained from mice immunized with different HCV-derived antigens were used in GrB activity assays (Supplementary Fig. S4). It was observed that splenocytes, CD4+, and CD8+ T cells from F or core immunized mice had slightly reduced GrB activity compared with CV immunized mice. The decrease in GrB enzyme activity in T cells obtained from mice immunized with F or core did not appear significantly lower than CV, possibly because of the lower limit of detection by spectrophotometer. The splenocytes and T cells obtained from NS3 immunized mice showed higher GrB enzymatic activity compared to F, core, or CV immunized mice, correlating with efficient induction of effector T cells and increased intracellular GrB expression against NS3. These results provide functional relevance to the observation of differential intracellular GrB expression.
Reduction in GrB expression in T cells obtained from F or core immunized mice correlates with reduced killing of peptide-loaded targets
In vitro target killing experiments were performed to determine the ability of T cells, with reduced expression of GrB, to kill peptide-loaded target cells. Splenocytes obtained from F, core, CV, or NS3 immunized mice were cultured for 5 days in vitro in the presence of peptide mixtures as described in the authors' flow cytometry experiments. The resultant effector cells were then incubated with peptide pool-loaded and CFSE-labeled EL-4 cells at an effector to target ratio of 30:1 for 4 h. It was observed that splenocytes obtained from F or core immunized mice had a significantly reduced ability to kill peptide loaded EL-4 cells compared to CV immunized mice (Fig. 4). The positive control splenocytes from NS3 immunized mice demonstrated significantly higher killing of peptide-loaded targets compared to CV immunized mice (Fig. 4). These results provide further functional confirmation that T cells obtained from mice immunized with HCV-derived F or core antigens have a reduced ability to kill target cells than do NS3 antigen primed T cells.
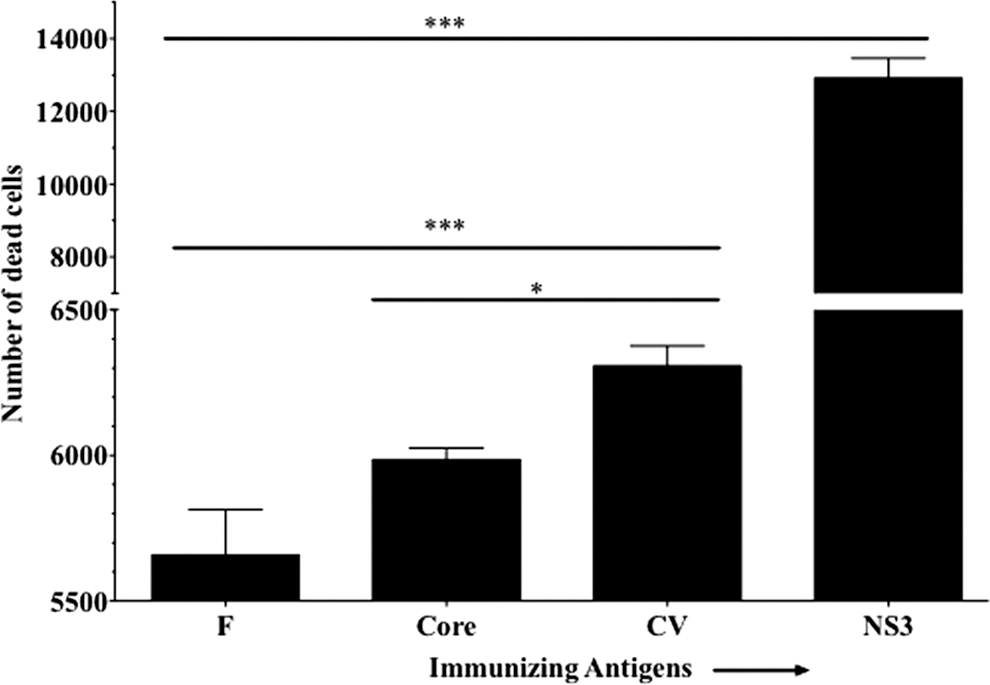
Splenocytes obtained from F or core immunized mice have lower ability to kill peptide-loaded EL-4 target cells. Cytolytic activity of splenocytes obtained from mice immunized with F, core, NS3, or CV was measured against EL-4 target cells pulsed with respective peptide mixtures. Dead cells (average cell counts 3,000/group) in peptide unloaded EL-4 target groups were subtracted from peptide-loaded targets to obtain antigen-specific killing. A 30:1 effectors-to-target ratio was used in a 4 h CFSE positive cells' loss assay. The data shown are average±S.D. from triplicate cultures and represent three repeated experiments. Statistical analysis was performed using one-way ANOVA. Differences were statistically significant at p<0.05 (denoted by *) and p<0.0007 (denoted by ***).
Addition of exogenous IL-2 partially reverses the decrease in GrB expression in splenocytes obtained from F or core immunized mice
IL-2 is necessary for proliferation, growth, and differentiation of T cells into effector cells. This study examined whether treatment with exogenous IL-2 in vitro could reverse the reduction in GrB expression in T cells from F or core immunized mice. Splenocytes obtained from F, core, or CV immunized mice were cultured with 1 μg/mL of their respective peptide mixtures with IL-2 for 5 days and examined for intracellular GrB expression in CD3+ CD4+ and CD3+ CD8+ T cells. In the presence of IL-2, upregulation of GrB expression was significant in CD4+ and CD8+ T cell populations from CV immunized mice (Fig. 5). T cells obtained from core or F immunized mice also demonstrated higher expression of GrB in the presence of IL-2 compared with cultures with no added IL-2, but GrB expression in the presence of IL-2 did not reach the levels observed in T cells from CV immunized mice (Fig. 5). These results suggest that IL-2 only partially reverses the decrease in GrB expression observed in the T cells of F or core immunized mice.
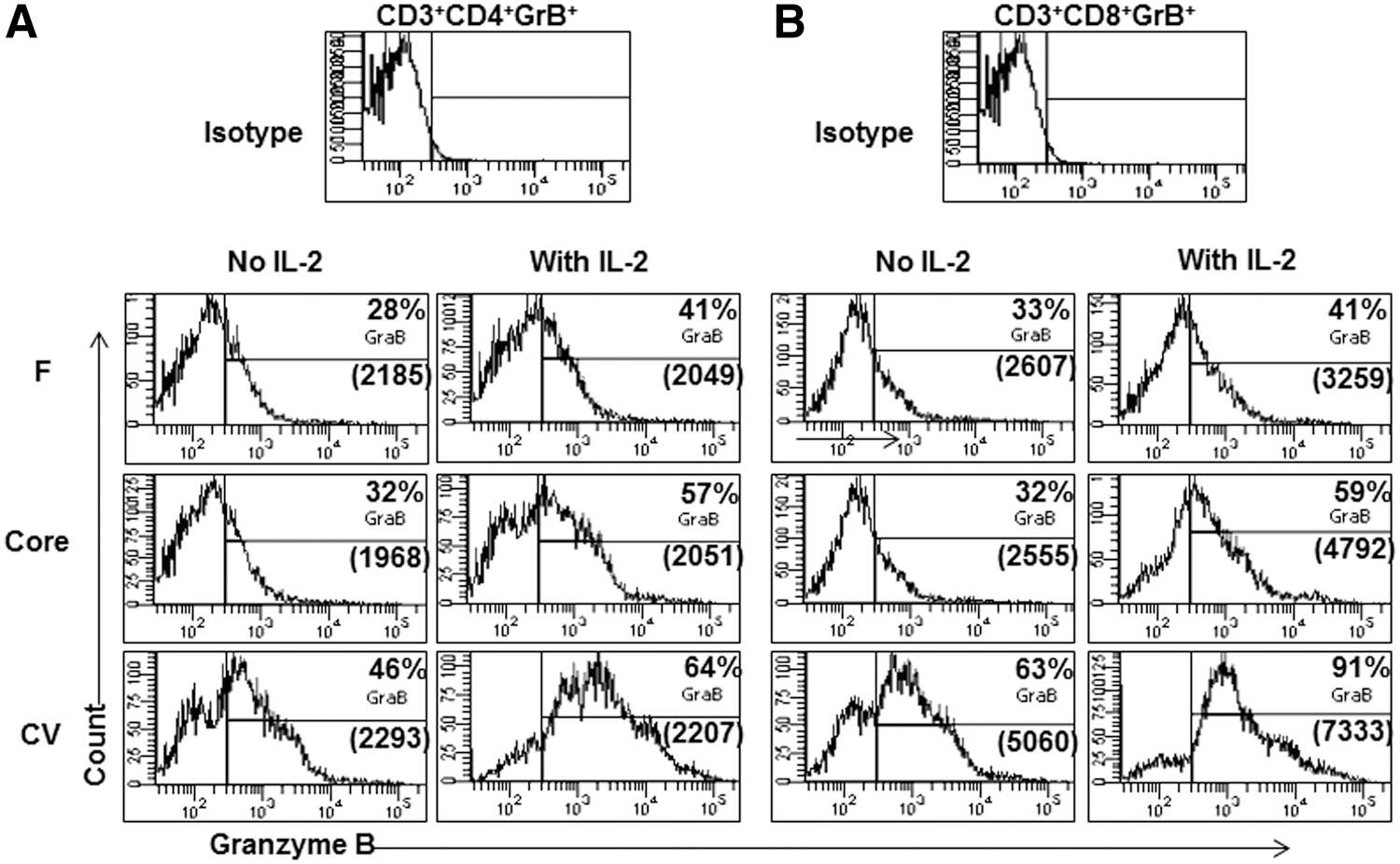
Addition of exogenous IL-2 to splenocytes partially reverses the GrB expression. Mice were immunized with recombinant adenovirus vectors (2×107/mouse) containing F, core, or no antigen CV. Splenocytes collected 8 days after immunization were cultured in the presence or absence of exogenous recombinant IL-2 (100 pg/mL) for 5 days in presence of peptide mixtures from respective proteins as described in Figure 3. Cells were then stained for extracellular CD3, CD4, CD8, and intracellular GrB. Gating was done as described in Figure 3. (
Modulation of GrB expression is NF-κB mediated
GrB expression is regulated through the NF-κB pathway in activated T cells, and IL-2 has been shown to activate transcription of GrB through NF-κB activation (37). In addition, it has been shown that there is an NF-κB binding site downstream of the GrB gene, which is responsible for GrB gene activation and which is functional in vitro and in vivo in natural killer (NK) and T cells (37). TLR agonists, for example double-stranded RNA binding through TLR-3, also activate NF-κB (1). Taken together, it can be reasoned that stimulation of TLRs in T cells can lead to NF-κB activation resulting in upregulation in GrB production. This study therefore examined whether T cells isolated from F or core immunized mice and cultured with mixtures of respective peptides in the presence or absence of IL-2, poly I:C, and/or PDTC (a known inhibitor of NF-κB) exhibited further reduction in GrB expression (Fig. 6). In the presence of PDTC, GrB expression was almost completely inhibited in both CD4+ and CD8+ T cells, even in the presence of strong NF-κB stimulators such as IL-2 and poly I:C (Fig. 6). These results indirectly suggest that NF-κB modulation may be involved in downregulation of GrB upon immunization with F or core containing recombinant adenovirus vectors. To test this possibility, mice with recombinant adenovirus vectors containing F, core, NS3, or CV (no antigen), along with a TLR-3 agonist poly I:C (Fig. 7), were immunized. Splenocytes obtained from the immunized mice were cultured for 5 days in vitro with mixtures of respective peptides at 1 μg/mL concentration, and GrB expression was examined in CD4+ and CD8+ T cells (Fig. 7). Intriguingly, the decrease in GrB expression was not only completely reversed in T cells upon co-immunization of mice with poly I:C, but GrB+ CD4+ and GrB+ CD8+ T cell levels were also significantly higher in both F and core immunized mice, and they showed killing of peptide loaded EL-4 target cells (data not shown), suggesting that functional effector T cells were induced in response to F and core antigens. It is noteworthy that the levels of GrB+ CD4+ and GrB+ CD8+ T cells in both F and core immunized mice were similar or higher than those of NS3 immunized mice (a positive control; Fig. 7). Mice immunized with F or core containing adenovirus vectors along with TLR-3 agonist poly I:C showed similar levels of proliferation (Fig. 8), and of IL-10 and IFN-γ production (Fig. 9) as the mice immunized with F or core containing adenovirus vectors only. However, profiles of some of the peptide-specific IL-10 production by T cell cultures from both F and core immunized mice were modulated upon co-immunization with poly I:C (Figs. 8 and 9).

When splenocytes obtained from immunized mice are treated with pyrrolidine dithiocarbamate (PDTC) in the presence or absence of IL-2 or poly I:C, GrB expression is abrogated. Mice were first immunized with recombinant adenovirus vectors containing F, core, NS3, or no antigen CV. Splenocytes obtained from immunized mice were cultured for 5 days in the presence of mixtures of peptides from respective proteins, as described earlier in Figure 3. They were also cultured with PDTC (a known inhibitor of NF-kB) in the presence or absence of IL-2 or poly I:C. Cells were stained and analyzed for intracellular GrB expression on (

Immunization of mice with recombinant adenovirus vectors containing F or core supplemented with TLR-3 agonist poly I:C reverses the downregulation of GrB expression. Mice were immunized once i.m. with recombinant vectors containing F, core, NS3, or CV along with poly I:C (20 μg/mouse). Eight days after immunization, splenocytes were harvested and cultured for 5 days in the presence of peptide mixtures from the respective proteins, as described earlier in Figure 3, followed by staining for extracellular CD3, CD4, CD8, and intracellular GrB. Gating was done using the strategy shown in Figure 3. (

Antigen-specific proliferation of T cells after a single i.m. immunization with F or core along with poly I:C. Peptide-specific proliferation of T cells after a single i.m. immunization of mice with 2×107 recombinant adenovirus vectors containing F or core and poly I:C (20 μg/mouse). Enriched T cells obtained from the splenocytes of immunized mice were cultured in vitro with irradiated syngeneic APCs in the presence of 1 μg/mL respective mixtures of overlapping peptides from F or core proteins for 5 days. The numbers on the x-axis of the graphs represent the peptide number described in Tables 1
–3. T cells and APCs incubated in the absence of peptides served as control. Cells cultured in the presence of comparable concentrations of DMSO were used as background to determine the stimulation indices (stimulation index=CPM obtained in the presence of peptides/CPM obtained in the presence of solvent DMSO control). (
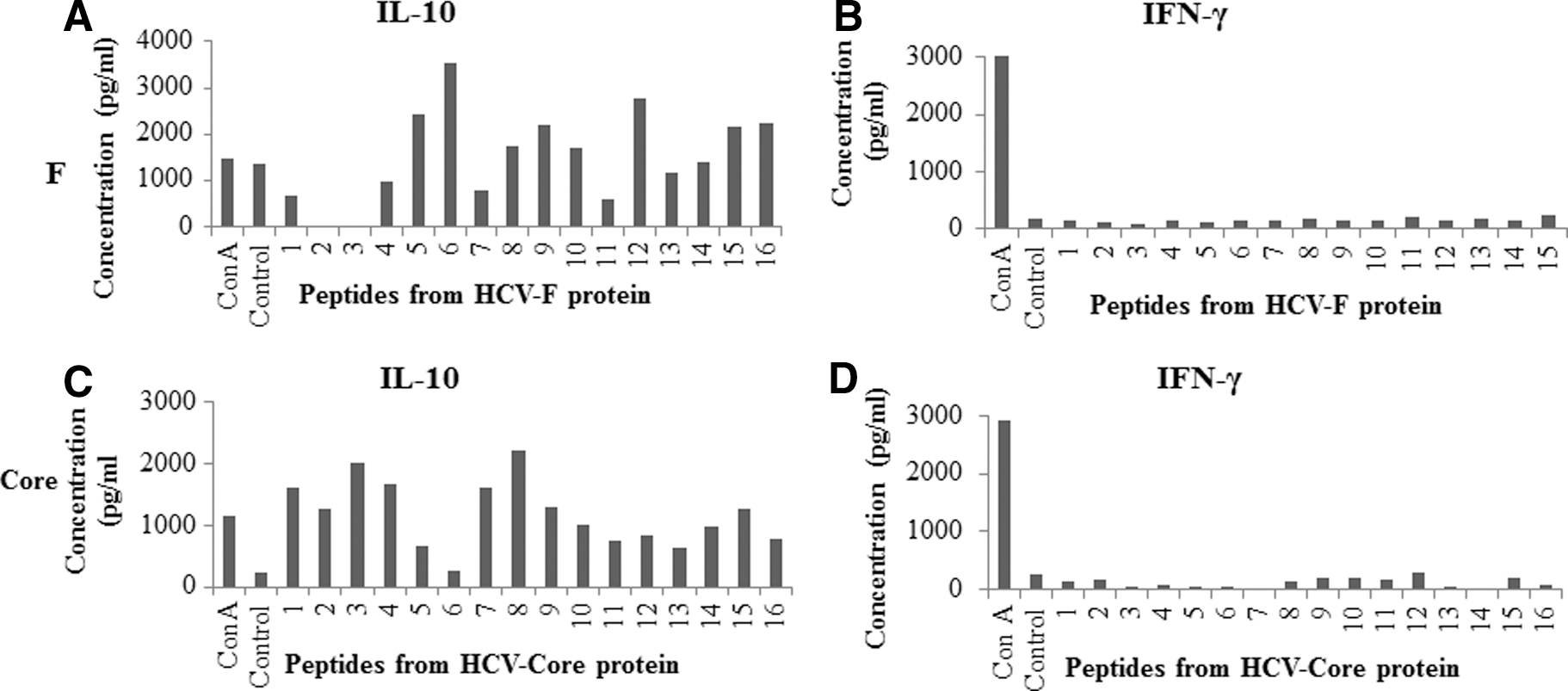
T cells obtained from mice immunized with F or core along with poly I:C produce high levels of IL-10 and very low or undetectable IFN-γ upon in vitro stimulation with F or core overlapping peptides. T cells obtained from spleens of mice immunized with F or core and TLR-3 agonist poly I:C were cultured with irradiated APCs and 1 μg/mL of respective peptides, as described in Figure 8. Supernatants from cultures were collected on day 4, and cytokines were analyzed by ELISA. Assays were done in duplicate, and average values are shown. Controls represent cultures without peptides. Con A represents the positive control for the assay. The numbers on the x-axis of graphs represent the peptide number, as described in Tables 1
–3. (
Discussion
HCV causes acute and chronic infection in humans. The virus is spontaneously cleared in 20–35% of infected patients, but persists in 65–80% of its victims due to insufficient innate and adaptive immune responses (48,70), and the rate of viral clearance is genotype dependent (49). The mechanisms of immune evasion by HCV have been investigated extensively (17,28,44,50,62) but are not yet clearly understood. This study reveals a new role of F and core proteins of HCV that might contribute to the inefficiency of T cell responses generated in chronic HCV infection: the disarming of effector CD4+ and CD8+ T cells' ability to kill infected target cells. Effector T cells provide antiviral immunity and clearance of viral infection by killing virus-infected target cells. To the authors' knowledge, they are the first to demonstrate downregulation of GrB as a possible mechanism to disarm effector T cells by a virus infection/antigen through this report and their previous publication (45). In an earlier report, whole irradiated or paraformaldehyde fixed parainfluenza virus type 3 was shown to inhibit GrB mRNA in human PBMCs and NK cells (40,69), but protein expression and the functional impact of the decrease in GrB mRNA were not studied. However, several other factors are involved in the T cells' ability to kill target cells. These include CD4+ T cell help for the effector function of CD8+ T cells in the form of antigen presentation and by expression of effector cytokines (40). Several viruses and/or viral antigens have also been known to interfere with the class I antigen processing and presentation machinery, leading to inefficient presentation of viral antigens for recognition by cytotoxic T lymphocytes (CTLs) (35,36).
Expression of co-inhibitory molecules such as PD-1 and CTLA-4 has been shown to be upregulated in chronic HCV infection, suggesting a mechanism of downregulation of T cell responses (45,63,73,74). However, these reports have mostly used peripheral blood T cells from chronic HCV-infected patients with long-term virus persistence, and may be more representative of T cell exhaustion due to continued antigen stimulation. The current findings of T cell modulation are distinct and unexpected in that after a single immunization with F or core antigens, even in the context of recombinant adenoviral vector, a substantial reduction in GrB expression was observed after 8 days. Since GrB expression was not determined specifically in peptide-specific T cells using tetramer staining, it is possible that bystander T cells also have reduced GrB expression in these mice. In contrast, another HCV antigen, NS3, in the context of the same adenovirus vector, induced T cells with greatly upregulated GrB production. However, co-administering the NS3 with F or core led to T cells with reduced GrB expression, suggesting that F and core have significant impact on the generation of effector T cells in response to NS3 and also that immunization with F or core antigens leads to reduced GrB expression in bystander T cells (Supplementary Fig. S2). Experiments were performed where GrB was determined right after harvesting the splenocytes from mice. In these experiments, the GrB levels were very low, and there was no significant difference in different groups (data not shown). This is not surprising in light of the literature from several mouse experiments as well as clinical trials (6,33,34,47,78,79,82). It is known that T cells need to be cultured in vitro with APCs and relevant antigen in order to allow proliferation, expansion, and enrichment of antigen-specific T cells. In clinical trials with various viral and cancer vaccines, antigen-specific CTL activity has invariably been shown with cells cultured in vitro for 4–5 days in the presence of antigen and autologous APCs (47,79,82). It is well-known that in vitro culture with specific antigens allows antigen-specific T cells to expand and/or be enriched, whereas non-specific T cells will die. A similar strategy has also been used here to allow expansion and enrichment of antigen-specific T cells. The culture technique would allow in vivo primed antigen-specific T cells to be restimulated, expand, survive, and be enriched in the presence of peptides derived from their respective proteins. It is possible that at different time points, differential regulation of GrB expression would be seen. Although significant proliferation upon stimulation of T cells with peptides from F or core antigens was not observed (Fig. 1), there was significantly more recovery of viable T cells after 5 days of culture from F, core, or CV immunized mice (33–40%) compared with unimmunized mice (2%). These results suggest antigen-specific T cells survived and were maintained in these cultures. The splenocytes from CV immunized mice were cultured in the absence of added antigen, and it is expected that culture of whole splenocytes allow APCs loaded with the adenovirus antigen to be carried into the culture for restimulation.
GrB has diverse roles in cellular immunity, including apoptosis of intracellular infected cells by caspase dependent and independent mechanisms (52), autoimmune destruction of pancreatic β cells in type 1 diabetes (72), effector T cell mediated clearance of tumors (10), and graft rejection (31). GrB positive CD4+ and CD8+ T cells have also been shown to have a significant role in antiviral immunity (2,3,43,68). HCV uses several strategies to evade the immune system, but a major mechanism is the modulation of T cell responses (62). Effectiveness of T cell activity in HCV infection could be compromised by lower expression of GrB upon priming of certain antigen-specific T cells. At this point, it is difficult to determine whether there is a real reduction in GrB expression, or whether there is simply no expansion of type 1 effector T lymphocytes in cultures from F and core immunized mice. However, the observation that the cultures from mice immunized with F or core had lower GrB+ T cells compared with control adenovector immunized mice suggests this is a result of immunization with F or core antigens. It is possible that intracellular levels of GrB appear to be downregulated because activated effector T cells have undergone degranulation. It has been reported that GrB expression can be altered due to different rates of degranulation in activated T cells (8). Cell surface expression of CD107b indicates recent cytotoxic cell degranulation, and has been used as a surrogate marker for GrB/perforin-mediated killing by effector T cells (8,84). However, the present results with CD107b expression (Supplementary Fig. S3) suggest this not to be the cause of GrB reduction in mice immunized with F or core.
The authors previously reported immune responses generated in mice immunized with adenovirus vectors containing HCV core or NS3 antigens. It was observed that antigen-specific splenic T cells from NS3 immunized mice proliferated strongly after secondary stimulation of recombinant NS3 protein or immunodominant peptides derived from NS3 (45). It was also demonstrated that immune-dominant peptides and recombinant core protein did not cause a proliferative response in T cells from core immunized mice (45). The current study examined proliferative responses of T cells obtained from F or core immunized mice against a series of overlapping peptides covering the entire core and F proteins (Fig. 1). Interestingly, none of the peptides stimulated significant T cell proliferation above background levels. Based on these results, one would assume that F and core antigens are simply not immunogenic in the H-2b background. However, when the supernatants of the peptide-stimulated cultures were examined for the presence of cytokines, it was surprising to find that significantly high levels of IL-10 were produced in response to several of the F and core peptides (Fig. 2), whereas IFN-γ, TNF-α, TGF-β, and IL-12 were not detected. In further experiments using intracellular cytokine staining, it was demonstrated that both CD4+ and CD8+ T cells produce IL-10 in an antigen-dependent manner (Supplementary Fig. S1). In an earlier study, the authors demonstrated high IL-10 production in T cells that were obtained from core immunized mice and then restimulated in culture with recombinant core protein (45). The present results corroborate these findings and extend them to specific peptides derived from core as well as from F protein, further establishing the antigen dependency and specificity of IL-10 production. It has been reported that HCV infection progresses to chronicity in intravenous drug users in the presence of high IL-10 and low IFN-γ expression (23). Excess IL-10 has the ability to suppress Th1 cytokine production and the proliferation of CD4+ and CD8+ T cells (5). This suggests that IL-10 may have a role in viral persistence and disease progression, with a low level required for immune regulation but higher levels detrimental for viral clearance (7,15,44).
The induction of a high percentage of Tregs in core immunized mice has been previously observed (46). The present study examined the expression of several negative regulatory markers (e.g., PD-1 and CTLA-4) on T cells. However, no significant difference was found between F, core, CV, and NS3 immunized mice (data not shown). In addition, unlike core immunized mice (45), F antigen immunized mice did not show an upregulation of Tregs (Supplementary Fig. S5). These results suggest that there may be different mechanisms of immune modulation in F or core immunized mice, as well as independent mechanisms of reducing GrB and inducing Tregs.
To determine the physiological significance of modulated GrB expression, GrB enzyme activity was examined in cell lysates by using a substrate cleavable by active GrB (Supplementary Fig. S4). Lysates were prepared from whole splenocytes or from purified CD4+ and CD8+ T cells obtained from F, core, CV, or NS3 immunized mice. Interestingly, the GrB enzyme activity was significantly higher in cell lysates from the NS3 group than in other lysates, suggesting the functional relevance of high intracellular levels of GrB in NS3 immunized mice. However, the enzyme activity in cell lysates from F or core groups was visibly lower than the CV group and highly significantly lower than the NS3 group. The enzymatic activity in F or core groups did not appear to be dramatically reduced compared to the CV group as expected from intracellular GrB expression. This could be explained by the workings of the colorimetric assay where an increase in absorbance by upregulation of a biological activity could be more easily quantified than downregulation, which results in a decrease in absorbance below the background level. Enzyme activities obtained in T cell lysates from both F and core groups were greatly reduced compared to NS3 lysates, which substantiates the observation and provides a physiological significance to downregulation of intracellular GrB expression.
Another parameter of functional significance in GrB expression is the potency of effector T cells in the killing of HCV-infected cells. EL-4 cells loaded with pools of peptides derived from F, core and NS3 antigens were used as targets and a CFSE assay was used to examine the reduction in the number of CFSE-loaded cells in the CTL assay (Fig. 4). It was observed that effector T cells generated from mice immunized with F or core have a lower ability to kill target cells than do effector T cells from CV or NS3 immunized mice, reflecting the lower expression of GrB in the F or core groups (Fig. 4). These results further substantiate the low ability of effector T cells from F and core immunized mice in killing infected target cells.
It has been reported that IL-2 treatment activates and enhances GrB expression in splenocytes (38). It was observed that addition of exogenous IL-2 enhanced the expression of GrB in splenocytes obtained from F or core immunized mice in both CD4+ and CD8+ T cells (Fig. 5). However, the percentage of cells expressing GrB was still lower than the percentage in splenocytes cultured in the presence of IL-2 from CV immunized mice, suggesting that exogenous IL-2 was only partially able to recover the expression of GrB. In addition, from this experiment, it is not clear whether the recovery of GrB expression is in antigen-specific T cells.
GrB expression is also regulated through NF-κB-mediated activation. It has been shown that the GrB gene has a downstream sequence that can bind NF-κB and activate GrB transcription (37). The NF-κB inhibitor PDTC (57) was used to examine if direct inhibition of NF-κB in T cells would lead to inhibition of GrB expression. Addition of PDTC to in vitro cultures of splenocytes led to very significant or almost complete inhibition of GrB expression in both CD4+ and CD8+ T cells obtained from F, core, CV, or NS3 immunized mice (Fig. 6). In addition, splenocytes cultured with IL-2 or poly I:C and treated with PDTC strongly inhibited GrB expression, suggesting that IL-2 or poly I:C stimulation cannot bypass the NF-κB mediated pathway of GrB regulation. These results indirectly support the notion that immunization of mice with F and core antigens could lead to downregulation of NF-κB activity that result in reduced expression of GrB. It is not clear whether it is the inhibition of NF-κB in T cells or in APCs that leads to modulation of GrB in T cells. NF-kB activation/phosphorylation in splenocytes of core/F immunized mice has not been quantified here, as it is a signaling event and may not be synchronized in all T cells and/or APCs. Several studies have reported that core and other HCV antigens inhibit NF-κB activation in DCs (39,44,61). In addition, a defect in NF-κB activation has been demonstrated in DCs obtained from patients chronically infected with HCV (61,85). However, direct inhibition of NF-κB activation in T cells has not been thoroughly studied. If NF-κB modulation does lead to a reduction in GrB expression in T cells obtained from mice immunized with F and core antigens, the mechanism of this modulation is not clear, and is being investigated in the authors' laboratory. To support the plausible mechanism of GrB reduction through modulation of NF-κB, it is reasonable to assume that TLR agonists, which are strong activators of NF-κB, might be able to reverse the reduction of GrB expression if injected together with adenovirus vectors containing F or core. Poly I:C, a TLR-3 agonist that represents double stranded RNA, was chosen (25). It was observed that immunization with adenovirus vectors containing F or core dramatically increased GrB expression in both CD4+ and CD8+ T cells when they were admixed with poly I:C (Fig. 7). Whether this reversal is by direct action on T cells or indirectly through DCs or other cells is not yet clear. In addition, there was no change in T cell proliferation (Fig. 8), or IFN-γ or IL-10 production (Fig. 9), implying that the observed effect of reversal of GrB expression is not likely due to induction of pro-inflammatory cytokines. TLR ligands appear to promote the capacity of DCs to induce T cell responses and are also known to act as adjuvants in the activation of antigen-specific T cells (42, 59). Synthetic TLR-3 ligands such as poly I:C have been known to act as potent immune adjuvants by enhancing DC cross-presentation and by promoting CD8+ T cell responses (66). However, this study is the first to show that poly I:C can reverse F or core-mediated dysfunction in effector T cells through reversing the suppression of GrB expression. It can be expected that agonists of other TLRs, by virtue of activating NF-kB, may exert a similar effect to reverse effector T cell dysfunction.
This work has uncovered a new strategy of immune modulation by HCV-derived F and core proteins, wherein modulation of GrB expression in effector T cells makes them less efficient in killing target cells. To the authors' knowledge, the downregulation of GrB by other viral antigens and the implications of this process for antiviral immunity have not been reported. This study opens new avenues in understanding T cell modulation by other pathogens. GrB expression is crucial for effective T cell responses in the clearance of virus-infected cells. However, at this point, it is not clear whether F and core proteins play a direct role in regulating GrB expression in T cells or whether these antigens stimulate T cells with an alternative effector phenotype that can influence the response or function of bystander T cells. Moreover, given the differences in MHC and T cell repertoires between mice and humans, a similar effect may or may not be observed in humans infected with HCV. These studies also suggest that adding a TLR-3 agonist as an adjuvant in a vaccine could effectively reverse the T cell dysfunction induced by F and core proteins and enhance effector T cell function. The findings are relevant to the design and investigation of prophylactic and/or therapeutic vaccines for chronic HCV infection. Further work is required to understand the mechanisms and signaling pathways involved in the modulation of GrB expression by direct and/or indirect effects of HCV antigens and their significance in HCV-infected individuals.
Footnotes
Acknowledgments
Technical support by J. Li and Dorothy Kratochwii-Otto (flow cytometry) is thankfully acknowledged. We gratefully acknowledge Dr. Mark Peppler for carefully reviewing and editing this manuscript. This work was supported by a grant from the Canadian Institutes of Health Research (MOP 79327) to BA. BA is a recipient of a senior scholar award from the Alberta Heritage Foundation for Medical Research (now Alberta Innovates Health Solutions).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.