
Abstract
The nucleocapsid (N) protein is the most conserved structural protein in equine arteritis virus (EAV). This study aimed to identify the minimal conserved B cell epitope on the EAV N protein. The purified N protein was used to immunize mice for preparing monoclonal antibody (mAb). The reactivity of mAb was evaluated by Western blot and immunofluorescence assay. Moreover, 11 overlapping peptides (named MBP-N1 to MBP-N11) were designed to localize the linear antigenic epitope within the N protein. The peptides were identified by indirect enzyme-linked immunosorbent assay (ELISA) and Western blot. The minimal conserved B cell epitope on the EAV N protein was identified. The homology analysis was also performed. An EAV N-reactive mAb was selected and designated as 1C11. Indirect ELISA results showed that overlapping domain between MBP-N10 and MBP-N11 was recognized by the mAb 1C11. Furthermore, the indirect ELISA and Western blot showed that 101QRKVAP106 was the minimal linear epitope of the EAV N protein. The homology analysis showed that the identified epitope was conserved among all EAV strains analyzed in this work, with the exception of the ARVAC. One EAV N-specific mAb (1C11) was developed, and a minimal linear peptide epitope (101QRKVAP106) within the N protein was identified.
Introduction
E
EAV is a positive-sense, enveloped, single-stranded RNA molecule with a length of 12.7 kb (22). It contains two large open reading frames (ORFs, 1a and 1b) and seven smaller ORFs (2a, 2b, and 3–7). ORFs 1a and 1b encode two replicase polyproteins (pp1a and pp1ab), whereas the ORFs 2a, 2b, 5, 6, and 7 encode the known EAV structural proteins E, GS (Gp2), GL (Gp5), M, and N, respectively. Moreover, ORFs 3 and 4 encode glycosylated membrane-associated proteins (Gp3, Gp4) whose functional role is still under debate (5,12). A genetic study has suggested that these protein expressions in EAV play important roles in the production of infectious progeny (26). Firth et al. (10) speculated that the N protein associated with Gp5-M heterodimer served as the structural core of the virion. Moreover, EAV N can be used as an alternative protein candidate of diagnostic antigens and accounts for 35–40% of the total virion protein (4). B cell epitopes are involved in the immune response against EAV (27). At the initial stage of infection, antibodies against N protein were produced first. Thus, it is particularly important to identify the precise epitope of the EAV N protein.
A previous study has indicated that the main N protein epitope is located within amino acid 1–69 residues (4). Zhao et al. (29) identified a completely conserved linear epitope that reacted with EAV-positive serum using Western blot. The present study aimed to identify a more precise B cell epitope with amino acid residues 1–106 using a monoclonal antibody (mAb) against EAV N protein.
Materials and Methods
Ethics statement
Care and use of laboratory animals and all animal experiments were in accordance with animal ethics guidelines established by the Institutional Animal Ethics Committee in China. All animal studies were approved by the Animal Ethics Committee of Harbin Veterinary Research Institute of the Chinese Academy of Agricultural Sciences (SYXK (H) 2006-032).
Cell lines and virus
SP2/0 myeloma and rabbit kidney 13 (RK-13) cells were cultured and maintained in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Carlsbad, CA) in a humidified 5% CO2 atmosphere at 37°C. All culture media were supplemented with 10% heat-inactivated fetal bovine serum (GIBCO, Invitrogen) and antibiotics (0.1 mg/mL streptomycin and 100 IU/mL penicillin; Sigma, St. Louis, MO). The Bucyrus strain of EAV (GenBank accession No. NC-002532.2, a highly cell culture–adapted strain provided by the key laboratory of Tropical and Subtropical Animal Viral Diseases in Yunnan province, China) was propagated in RK-13 cells and stored at −80°C.
Expression and characterization of recombinant EAV N protein
The full-length sequence of EAV N gene was cloned by reverse transcription polymerase chain reaction (RT-PCR) using the following primers: 5′-CCC GGA TCC ATG GCG TCA AGA CGA TC-3′ (upstream) and 5′-TTT GTC GAC TTA CGG CCC TGC TGG AGG CGC AAC-3′ (downstream). The primers contained BamH I and Sal I restriction sites (italicized). The purified and digested PCR product was ligated into an expression vector pET32a (Novagen, Darmstadt, Germany). The pET-N recombinant plasmid was transformed into Escherichia coli BL21 (DE3), and a final concentration of 1 mM isopropyl-β-D-1-thiogalactopyranoside (IPTG, Invitrogen) was used for inducing the expression of N protein. The cultures were centrifuged at 7,000 g for 10 min, and then lysed by sonication with phosphate buffered saline (PBS, pH 7.4). Finally, the recombinant N protein fused with 6 × His-tags was purified by Ni affinity chromatography according to the manufacturer's instruction (Invitrogen) and evaluated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot with the polyclonal anti-EAV serum. A horseradish peroxidase (HRP)-labeled rabbit antihorse IgG (1:40,000; Sigma) was used as a secondary antibody. H2O2 was used as the substrate and 3,3-diaminobenzidine (DAB; Sigma) was used to visualize the results.
Preparation and characterization of mAbs against N protein
EAV N-reactive mAb was generated as previously described (30). Briefly, 6-week-old female BALB/c mice were immunized with the purified His-tag-N protein (100 μg per mouse) mixed with an equal volume of Freund's complete adjuvant (FCA, Sigma). Two booster injections containing the same amount of purified His-tag-N protein in an equal volume of Freund's incomplete adjuvant (FIA) were given at 2-week intervals. The purified N protein without adjuvant was injected intraperitoneally as the final immunization. After 3 days, the mice were euthanized, and their splenocytes were fused with SP2/0 myeloma cells using polyethylene glycol (PEG4000, Sigma). The hybridoma cells were seeded into 96-well plates and selected in hypoxanthine-aminopterin-thymidine (HAT, Sigma) selection medium. After 5 days, the medium was removed and replaced with hypoxanthine-thymidine (HT)-DMEM medium. After selection in HAT and HT medium, hybridoma supernatants were screened for evaluating reactivity and specificity of mAb by Western blot and immunofluorescence assay (IFA). Briefly, hybridoma supernatant of mAb (1:2) was utilized as the primary antibody. The HRP-conjugated goat antimouse IgG (1:40,000) and the fluorescein isothiocyanate (FITC)-labeled antimouse antibody IgG (1:200) were employed as the secondary antibody for Western blot and IFA, respectively. The class and subclass of the mAb was determined using a SBA ClonotypingTM System/HRP (Southern Biotechnology Associates, Inc., Birmingham, AL).
Polypeptides design and expression
Eleven overlapping peptides spanning the N protein were designed (Table 1) in order to localize the linear antigenic epitope. For each peptide, a pair of oligonucleotide strands was synthesized. The production of each pair of oligonucleotide strands was cloned into the BamH I and Sal I sites of pMAL™-C4x vector and expressed as MBP-N fusion proteins. These MBP-fused proteins were named consecutively MBP-N1 to MBP-N11 (Table 1). In brief, the recombinant plasmids were transformed into E. coli Rosetta (DE3; Novagen, San Diego, CA), and IPTG was used to induce protein expression. Furthermore, in order to identify the minimal linear peptide epitope, seven shorter MBP-N11—namely, MBP-N11-1, MBP-N11-2, MBP-N11-3, MBP-N11-4, MBP-N11-5, MBP-N11-6, and MBP-N11-7—were constructed by continuously deletion amino acid from N and C terminal (Table 2). The MBP-N-fusion proteins were used to detect mAb by indirect enzyme-linked immunosorbent assay (ELISA) and Western blot.
ELISA
Ninety-six-well plates were coated with MBP-N proteins in 0.1 M carbonate buffer (pH 9.6) at 4°C overnight and blocked with 5% skim milk for 1 h at 37°C. After washing three times with PBS plus 0.5% Tween-20 (PBST), 100 μL hybridoma supernatant was added to wells and incubated at 37°C for 1 h. Then, the plates were washed three times with PBST and incubated with diluted HRP-conjugated goat antimouse IgG (1:40,000; Abcam, Cambridge, United Kingdom) at 37°C for 1 h. The color was developed with 3,3′-5,5′-tetramethyl benzidine (TMB, 100 μL), and the reaction was stopped with 2 M H2SO4. The absorbance at 450 nm was measured. The average from triplicate assays was calculated. The MBP tag without a fused polypeptide was used as a control for coating efficiency.
Homology analysis
To evaluate the conservation of the identified linear epitope among EAV from different geographic areas, the identified epitope and the corresponding regions of other regional EAV virus strains were aligned using DNAMAN software v5.2.2.0 (Lynnon BioSoft Inc., Los Angeles, CA).
Results
Production of recombinant EAV N protein and mAb
As shown in Figure 1A, EAV N protein was successfully expressed using the procaryotic expression system. A clear band with expected molecular weight (34 kDa) was displayed. Accordingly, the recombinant EAV N protein was suitable as an antigen for immunization and hybridoma screening.
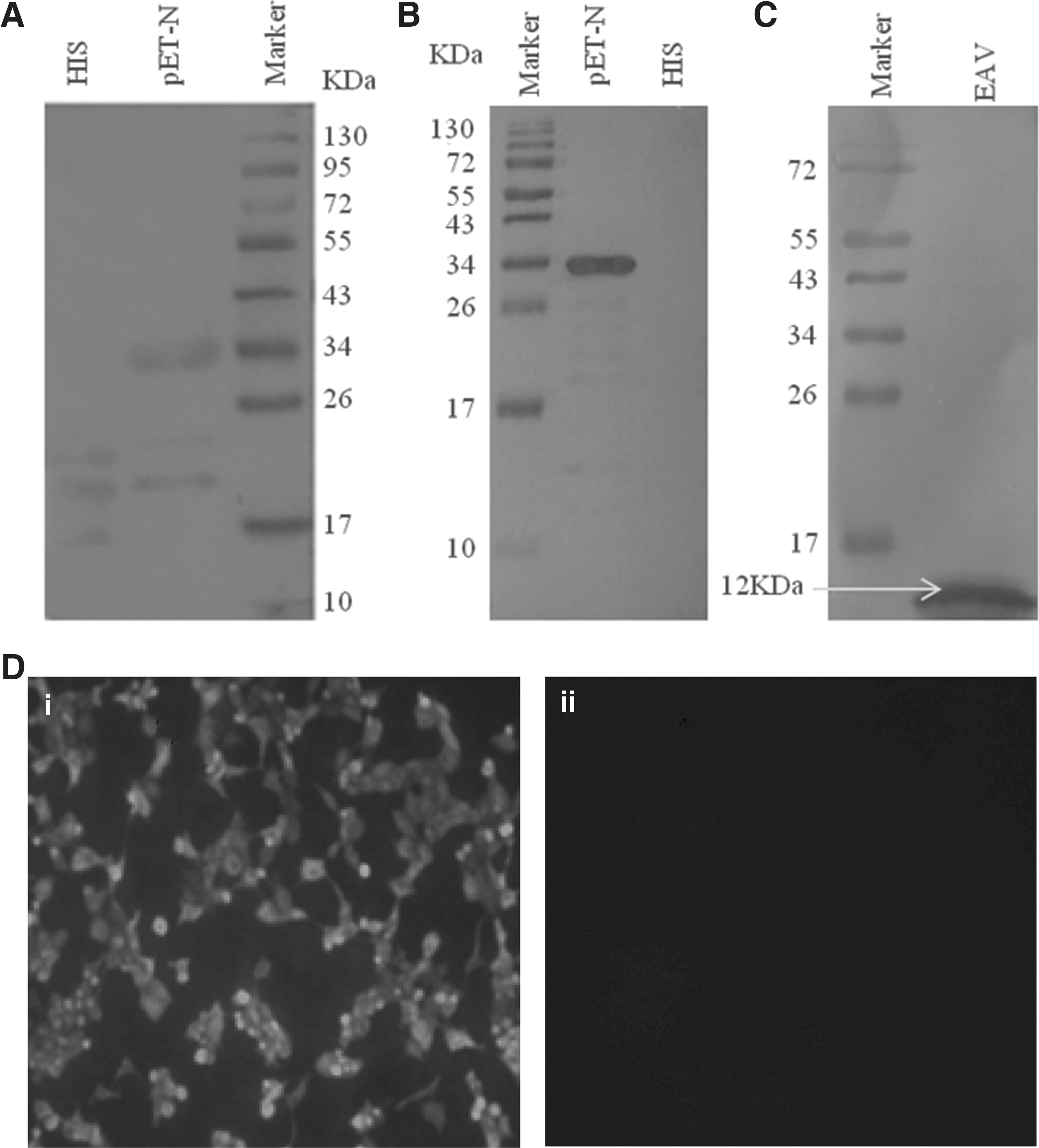
The expression of equine arteritis virus (EAV) N protein and the reactivity analysis of monoclonal antibody (mAb) 1C11.
After cell fusion and selection, an EAV N-reactive mAb generated from one hybridoma cell line was selected for its strong reactivity against N protein. This mAb was designated as 1C11. As shown in Figure 1B and C, mAb 1C11 reacted well with recombinant EAV N protein and the native EAV protein (12 kDa). The reactivity of mAb in RK-13 cells was also assessed by IFA (Fig. 1D). As shown, the mAb only reacted with EAV infected RK-13 cells.
Identification of the minimal linear peptide epitope
The results of indirect ELISA showed that only MBP-N10 (91TVSWVPTKQIQRKVAP106) and MBP-N11 (95VPTKQIQRKVAPPAGP110) were respectively recognized by the mAb 1C11 (OD450 >1; Fig. 2A). All the other fragments (MBP-N1-9) failed to react with the mAb 1C11. Moreover, there were 12 overlapping amino acids (95VPTKQIQRKVAP106) between MBP-N10 and MBP-N11. Therefore, a series of truncated polypeptides were expressed as MBP-fusion proteins to identify the minimal linear peptide epitope within this overlapping domain (Table 2). Ultimately, the indirect ELISA showed that N11-1-6 could be recognized by the mAb 1C11, whereas N11-7 could not (Fig. 2B). Western blot showed the same result (Fig. 2C). In other words, N11-6 (101QRKVAP106) was the minimal linear epitope for the reactivity of the EAV N protein recognized by mAb 1C11.
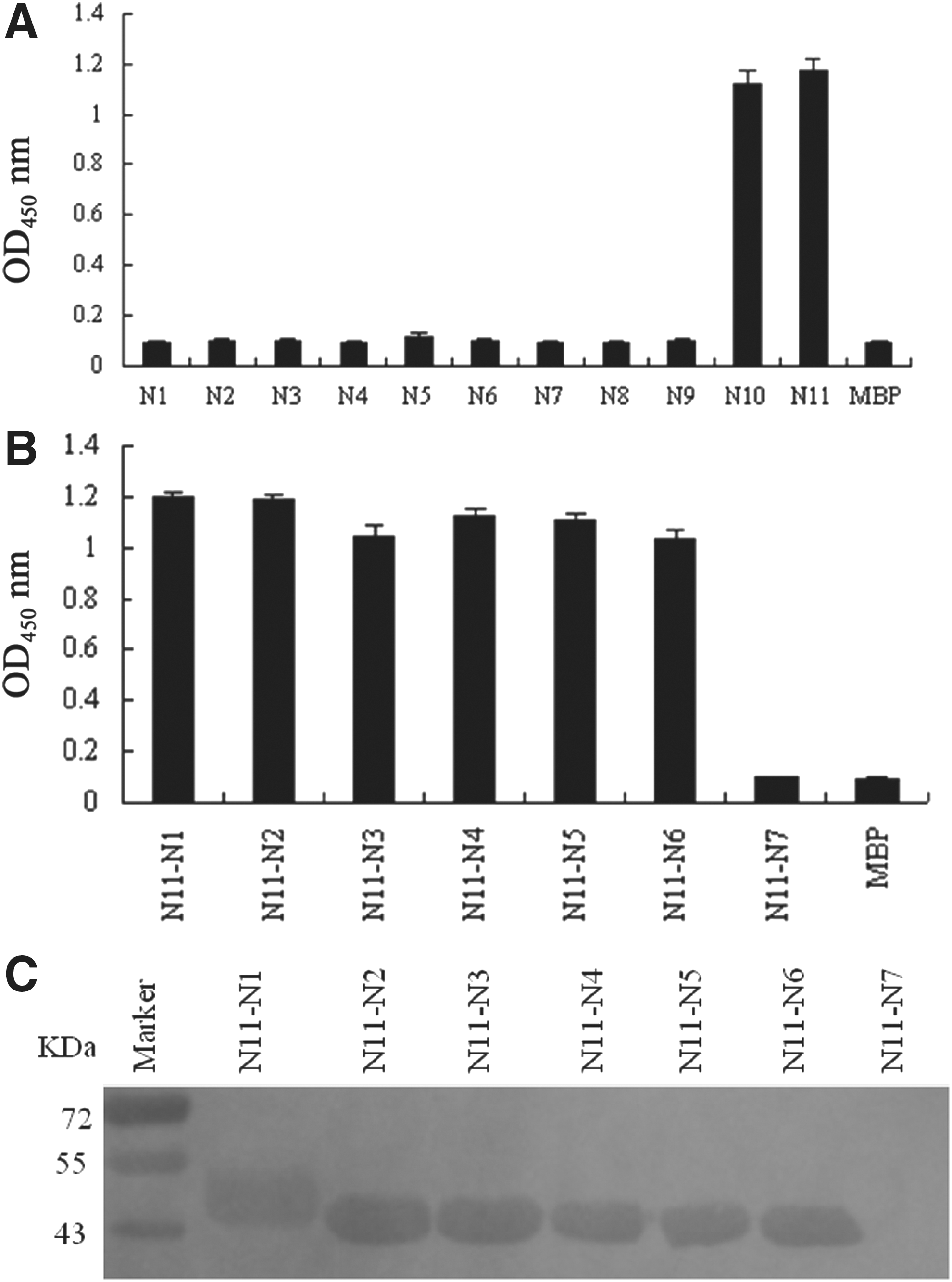
A total of 11 overlapping peptides encompassing the entire sequence of the EAV N protein were expressed using mAb 1C11 by enzyme-linked immunosorbent assay (ELISA).
Homology analysis
Once the minimal linear epitope (101QRKVAP106) was identified, sequence alignment was performed to evaluate the conservation of the identified epitope among 30 different regional EAV viruses. It was found that the identified epitope as framed in box in Figure 3 was conserved among all EAV strains analyzed in this work, with the exception of the ARVAC, which is a modified live virus vaccine strain.

Sequence alignment of the identified minimal linear epitope (101QRKVAP106) of EAV N protein in 30 strains.
Discussion
Mapping location of viral protein epitopes and defining the degree of their conservation plays an important role in understanding of the antigenic structure, virus–antibody interactions, vaccine design, and clinical applications. In this study, the B cell epitopes of EAV N protein were analyzed using a mAb, and a precise epitope (101QRKVAP106) was determined.
It has been indicated that the N protein is a good candidate antigen for the diagnosis of serological disease. Several mAbs directly against the N protein have been characterized and evaluated by indirect ELISA and IFA techniques. Kondo et al. (15) found six mAbs can precipitate the N protein, which provides the first evidence for the presence of EAV antigen during the initial phase of infection. Weiland et al. (25) identified 10 mAbs against EAV N protein, but they found that 10 virus isolates reacted with only one of the two mAbs (mAb E6/E3 and mAb E7/d15-c1), which had the highest neutralization titers. Besides, Starick et al. (23) produced a mAb against the N protein to detect EAV, and an antibody dilution of 1:60 was chosen as the standard to achieve the maximum brilliance and the highest reliability for routine diagnosis. Recently, a mAb named 2B1 was also developed using recombinant N protein expressed in E. coli (29). In the present study, another mAb named 1C11 against EAV N protein was identified, and it reacted well with EAV in Western blot and IFA. However, whether this antibody can be used as a useful detection tool in EAV diagnosis is still unclear.
Epitope mapping using mAbs has become a powerful tool to study protein structure and provides new tools to diagnose diseases and design vaccines (28). mAbs are useful and effective for mapping antigenic epitopes of viral proteins. Moreover, the peptide ELISA for EAV diagnosis has been developed by Metz et al. (19) using serum samples that were previously analyzed by virus neutralization, and they demonstrated that the indirect ELISA was more rapid and simple in quantification of EAV antibodies. In the present study, indirect ELISA was performed, and 101QRKVAP106 was defined as the minimal linear epitope of EAV N protein using mAb 1C11. The present result differs from the previous study, which identified 38KPPAQP43 as the minimal requirement for 2B1 recognition (29). Compared to the previous study, some similarities were observed. First, the open reading frame 7 (ORF7) gene, encoding EAV N protein, was amplified to construct the recombination protein both in the previous study and the current study. Second, the immunized animals were 6-week-old female BALB/c mice in both studies. Therefore, it is speculated that differential mAbs identified by distinct authors contribute to different epitopes, and more studies may be needed to determine which mAb is better.
To the authors' knowledge, no previous reports about the identified epitope 101QRKVAP106 on the N protein have been published. In addition, the identified epitope 101QRKVAP106 only contains six amino acids, which might be useful as a molecular marker in scientific research. However, the current antibody is isolated from mice, and it is not known whether a similar epitope would be recognized by an antibody generated in horses, the natural host of EAV. Therefore, future studies are still needed.
In addition, the crystal structure of the EAV-NΔ48 protein has been analyzed by Deshpande et al. (6). The structure consisted of three α-helices, two β-sheets, and shorter C- and N-terminal helices. This structure is similar to that of PRRSV-NΔ57 (7). According to the crystal structure, the identified six amino acids (KPPAQP) are located at the α3 helix, which has the greatest mobility. Moreover, sequence alignment showed that the identified epitope was very conservative among distinct regional EAV strains, but with a mutation of one amino acids on the ARVAC N protein epitope. This result suggests a slight regional difference emerged in this epitope. Therefore, it is possible to distinguish anti-Bucyrus EAV antibody from anti-ARVAC EAV antibody by using the epitope as antigen.
Conclusions
In conclusion, one EAV N-specific mAb (1C11) was developed and a minimal linear peptide epitope within the N protein (101QRKVAP106) was identified. However, future studies should be carried out to evaluate the potential application of the identified epitope for vaccine.
Footnotes
Acknowledgments
This work was supported by the Science Technology Research Project of Heilongjiang Provincial Education Department, Fund Number 12531386.
Author Disclosure Statement
No competing financial interests exist.