
Abstract
Human immunodeficiency virus type 1 (HIV-1) envelope glycoprotein 120 (gp120) has been reported to be toxic to the hippocampal neurons, and to be involved in the pathogenesis of HIV-1-associated neurocognitive disorders (HAND). Accumulating evidence has demonstrated that voltage-gated potassium (Kv) channels, especially the outward delayed-rectifier K+ (Ik) channels, play a critical role in gp120-induced cortical neuronal death in vitro. However, the potential mechanisms underlying the hippocampal neuronal injury resulted from gp120-mediated neurotoxicity remain poorly understood. Using whole-cell patch clamp recording in cultured hippocampal neurons, this study found that gp120 significantly increased the outward delayed-rectifier K+ currents (Ik). Meanwhile, Western blot assay revealed that gp120 markedly upregulated Kv2.1 protein levels, which was consistent with the increased Ik density. With Western blot and terminal deoxynucleotidyl transferase dUTP nick end labeling assays, it was discovered that gp120-induced neuronal injury was largely due to activation of Kv2.1 channels and resultant apoptosis mediated by caspase-3 activation, as the pharmacological blockade of Kv2.1 channels largely attenuated gp120-induced cell damage and caspase-3 expression. Moreover, p38 MAPK was demonstrated to participate in gp120-induced hippocampal neural damage, since p38 MAPK antagonist (SB203580) partially abrogated gp120-induced Kv2.1 upregulation and neural apoptosis. Taken together, these results suggest that gp120 induces hippocampal neuron apoptosis by enhancement of the Ik, which might be associated with increased Kv2.1 expression via the p38 MAPK pathway.
Introduction
H
HIV-1 virus does not directly infect neurons themselves and cause the neurological symptoms (10,17,23). Indeed, increasing evidence has suggested that neuronal apoptosis in HAND is induced by indirect effects of multiple soluble neurotoxic factors released from virion and HIV-1-infected macrophages/microglial cells, such as quinoline, arachidonic acid, nitric oxide, platelet activating factor, matrix metalloproteinase, cytokines, chemokines, and HIV-1 virus envelope protein glycoprotein 120 (gp120) (2,7,10). Among all these factors, gp120 is considered one of the primary pathogens that lead to the neuronal injury and death in HAND patients (5,36). However, the potential mechanisms of gp120-induced neurotoxicity are not fully understood.
It is well known that apoptosis is characterized by cell volume decreases, caspase activation, and DNA fragmentation in a variety of cell types. Accumulating data have demonstrated the involvement of K+ homeostasis in these processes (4,33), and the role of voltage-gated potassium (Kv) channels in the apoptotic pathways mediated by gp120 has been of considerable research interest.
Numerous Kv channels with distinct localizations and properties are expressed throughout the CNS, where they contribute to neuronal cell membrane excitability and synaptic transmission (39). Henceforth, Kv channels have been characterized extensively in the mammalian hippocampus, which is closely associated with information processing and cognitive functions (14). Kv2.1 has been considered as a major subtype of the Kv channels in neurons and plays a critical and dynamic role in regulating the transmission of electrical signals in multiple neuronal damage models (6,25). Under the condition of serum deprivation, the extensive and constitutive dephosphorylation of Kv2.1 can be activated. Then, abundant Kv2.1 transfer to the cell membrane, which increases the current density of Kv2.1 and K+ efflux (20). Increasing evidence has shown that mitogen-activated protein kinases (MAPKs) are involved in several biological activities, including differentiation, proliferation, apoptosis, and inflammation (30). Several lines have demonstrated that the apoptotic surge of potassium currents is mediated by p38 phosphorylation of Kv2.1 (31,32). However, the mechanisms of the involvement of p38 MAPK and Kv2.1 in gp120-induced neuronal damage remain to be determined.
The present study hypothesizes that gp120 induces hippocampal neuronal apoptosis by enhancement of Kv channel functions through p38 MAPK phosphorylation. The results demonstrate that gp120 significantly enhances the Ik, leading to hippocampal neurotoxic activity. These adverse effects can be partially abolished by Kv2.1 channel blocker GxTX-1E. Furthermore, the antagonism of p38 MAPK signaling pathway also ameliorates gp120-mediated neurotoxic activity.
Materials and Methods
Animals
All experiments involving animals and tissue samples were conducted in accordance with the guide for the Care and Use of Laboratory Animals of the National Institutes of Health (NIH, USA), and all procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Nanjing Medical University (China; Permit Number: 20110521). Pregnant Sprague-Dawley rats were purchased from the Animal Center of Jiangsu Province, Nanjing, China (SCXK [Su] 2002-0031). All animals were maintained in independent ventilation cages at an ambient temperature of 23 ± 1°C and humidity of 55 ± 5% with a 12 h light/dark cycle. They were allowed access to diet and tap water ad libitum.
Primary culture of rat hippocampal neurons
Purified hippocampal neurons were prepared from rat embryos as described previously (26). Briefly, female Sprague-Dawley rats with 18–19 days of gestation were anesthetized, and embryonic pups were surgically removed. The hippocampi were harvested and digested using 0.25% trypsin for 15 min at 37°C. After washing in HBSS, tissue mixtures were centrifuged, decanted, and sequentially passed through a 100 μm and 40 μm mesh. The cells were seeded at a density of 2 × 106/dish in poly-D-lysine-coated 60 mm culture dishes, 2 × 105/dish in poly-D-lysine-coated 35 mm, and 1.5 × 105/well in poly-D-lysine-coated 24-well plates and maintained in neurobasal medium (Invitrogen) supplemented with 2% B-27 serum-free supplement, 1% penicillin/streptomycin, 0.2% fetal bovine serum, and 0.25 mM L-glutamine (Invitrogen) for 7–10 days. The purity of hippocampal neural cells was determined by staining with microtubule-associated protein-2 (MAP-2, a mature neuronal marker) antibody (Millipore), and >90% of MAP-2 positive cells were obtained.
Electrophysiological techniques
To measure outward K+ currents of the hippocampal cells, the whole-cell patch clamp recording techniques was used with an Axopatch 200B amplifier (Molecular Device), Patch electrode (4–8 MΩ) made from borosilicate (WPI, Inc.) with a P-97 micropipette puller (Sutter Instruments). The pipette solution for voltage-clamp experiments filled with (in mM): 120 potassium gluconate, 10 KCl, 5 NaCl, 1 CaCl2, 2 MgCl2, 11 EGTA, 10 HEPES, 2 Mg-ATP, and 1 GTP (pH adjusted to 7.3 with KOH). The extracellular solution contained (in mM): 140 NaCl, 5 KCl, 2.5 CaCl2, 10 HEPES, 10 glucose, and 0.001 tetrodotoxin (TTX; Tocris; pH adjusted to 7.3 with NaOH). After establishment of the whole-cell patch configuration, the cells were allowed to stabilize for 3–5 min for tests. Conventional capacitance and series resistance compensation routines were employed. The cell membrane capacitance measured from each cell was utilized for computing the whole-cell current density. Junction potential pipette resistance was corrected, and cell capacitance was compensated (∼80%). Current signals were filtered at 1 kHZ and digitized at 5 kHZ using a Digidata 1440A digitizer (Molecular Devices). The current and voltage traces were displayed and recorded using a pClamp10.2 data acquisition/analysis system (Molecular Devices).
MTT assay
Cell viability was measured by MTT (3-[4,5-Dimethyl-2-thiazolyl]-2,5-diphenyl − 2H-tetrazolium bromide; Sigma) assay at diverse concentration of gp120 (100, 200, 500, 1,000, and 2,000 pmol/L). Hippocampal neurons were seeded at a density of 8 × 104 cells/well in 48-well plates. Cell viability was measured after 24 h of incubation. The media were replaced by 400 μL of neurobasal containing 40 μL MTT (5 mg/mL) to each well. After 4 h, the culture medium was removed, and the crystals formed were dissolved by adding 300 μL dimethylsulfoxide (DMSO; Merck) per well. The absorbance at 490 nm was measured with a TECAN microplate reader. The experiment was repeated three times.
Terminal deoxynucleotidyl transferase dUTP nick end labeling assay
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining (Roche Applied Science) was performed to evaluate apoptotic neurons by using an in situ cell death detection kit. Briefly, rat hippocampal neurons were grown on poly-D-lysine coated coverslips at a density of 1.5 × 105/well in 24-well plates, and the cells were pretreated with Kv2.1 blocker, GxTX-1E (Peptides International, Inc.) for 30 min before adding 1,000 pM gp120 (Sino Biological, Inc.). After 24 h, neurons were fixed with 4% paraformaldehyde and permeabilized 0.1% Triton X-100 in 0.1% sodium citrate solution for 2 min on ice. Cells were then incubated with the TUNEL reaction mixture containing terminal deoxynucleotidyl transferase and fluorescein-labeled nucleotides at 37°C for 60 min. After the final wash, coverslips were mounted in VECTASHIELD Mounting Medium with DAPI (Vector Laboratories, Inc.) and visualized by fluorescent microscope using a 40× objective. Apoptotic cells were determined by counting percentage of positive-stained cells in 12 randomly selected fields.
Western blot analyses
Total proteins of hippocampal neurons were suspended in RIPA buffer (Sigma-Aldrich) containing protease inhibitor cocktail (1:500; Sigma-Aldrich) for 30 min on ice. They were then centrifuged at 12,000 g for 10 min at 4°C. The clarified lysates were transferred to fresh tubes on ice for analysis. Total proteins of 20 μg were separated by electrophoresis on 10–12% Tris-HCl gels and transferred to polyvinylidene difluoride membranes. Membranes were blocked with 5% dry milk in Tris-buffered saline (TBS; all from Bio-Rad Laboratories) and probed overnight at 4°C with primary Abs including rat Monoclonal antibody (1:500; NeuroMab), phospho-p38 MAPK, p38 MAPK, phospho-ERK1/2, ERK, phospho-JNK, JNK (1:1,000; Cell Signaling Technology), caspase-3 (1:1,000; Cell Signaling Technology), Kv2.2 (1:100; Santa Cruz), and antimouse β-actin monoclonal antibody (1:5,000; Sigma-Aldrich). Membranes were washed (four times, 10 min each) in TBS with 0.2% Tween (TBS-T) and incubated with HRP-conjugated antirabbit or antimouse secondary antibody (1:10,000, Jackson ImmunoResearch Laboratories) for 2 h at room temperature. Labeled proteins were visualized by Pierce ECL Western blotting substrate (Thermo Scientific). Band densities of phospho-p38 were normalized to total p38 in each sample.
Isolation of total RNA and real-time polymerase chain reaction
Hippocampal neurons were treated with the indicated concentrations of gp120 for 12 h, and total RNA was extracted by using TRIZOL (TaKaRa Bio, Inc.) according to the manufacturer's instructions. One microgram of RNA was reverse-transcribed using PrimeScriptTM RT Master Regent (TaKaRa Bio, Inc.). Then, cDNA was amplified by polymerase chain reaction (PCR) using specific primer Kv2.1 (267 bp; forward 5′-ACA CCA TCA CCA TCT CTC AAG G-3′ and reverse 5′-CTA ATT GTC AGC TCA-CCC CGA-3′) according to the manufacturer's instructions for SYBR Premix Ex TaqTM (TaKaRa Bio, Inc.). The temperature profile was as follows: initial denaturation at 95°C for 30 sec, 40 cycles of PCR step at 95°C for 5 sec, and then 60°C for 31 sec. β-actin was used as an internal control to evaluate relative expression of Kv2.1.
Statistical analysis
All data were expressed as mean ± standard error of the mean and graphed using GraphPad Prism v5 and Origin v8.0 software (OriginLab). Statistical analyses were performed by one-way analysis of variance or by q tests (SNK). Statistical significance was assumed at p < 0.05.
Results
Enhancement of outward delayed-rectifier K+ currents (Ik) by gp120 in cultured rat hippocampal neurons
To determine the effects of gp120 on Ik, 4-AP was initially used to block the transient outward K+ currents (IA), thereby sparing the Ik. The whole-cell outward K+ current was evoked by a depolarizing pulse +80 mV in the presence of 5 mM 4-AP, while the initial holding potential was set to −80 mV. The data showed that 200 pM gp120, which is used in the study by Chen (5), dramatically increased the average instantaneous K+ current density (74.1 ± 9.4 pA/pF, n = 10) compared with the control group (57.5 ± 5.4 pA/pF, n = 9; p < 0.05), suggesting an enhancement of gp120 on Ik (Fig. 1).
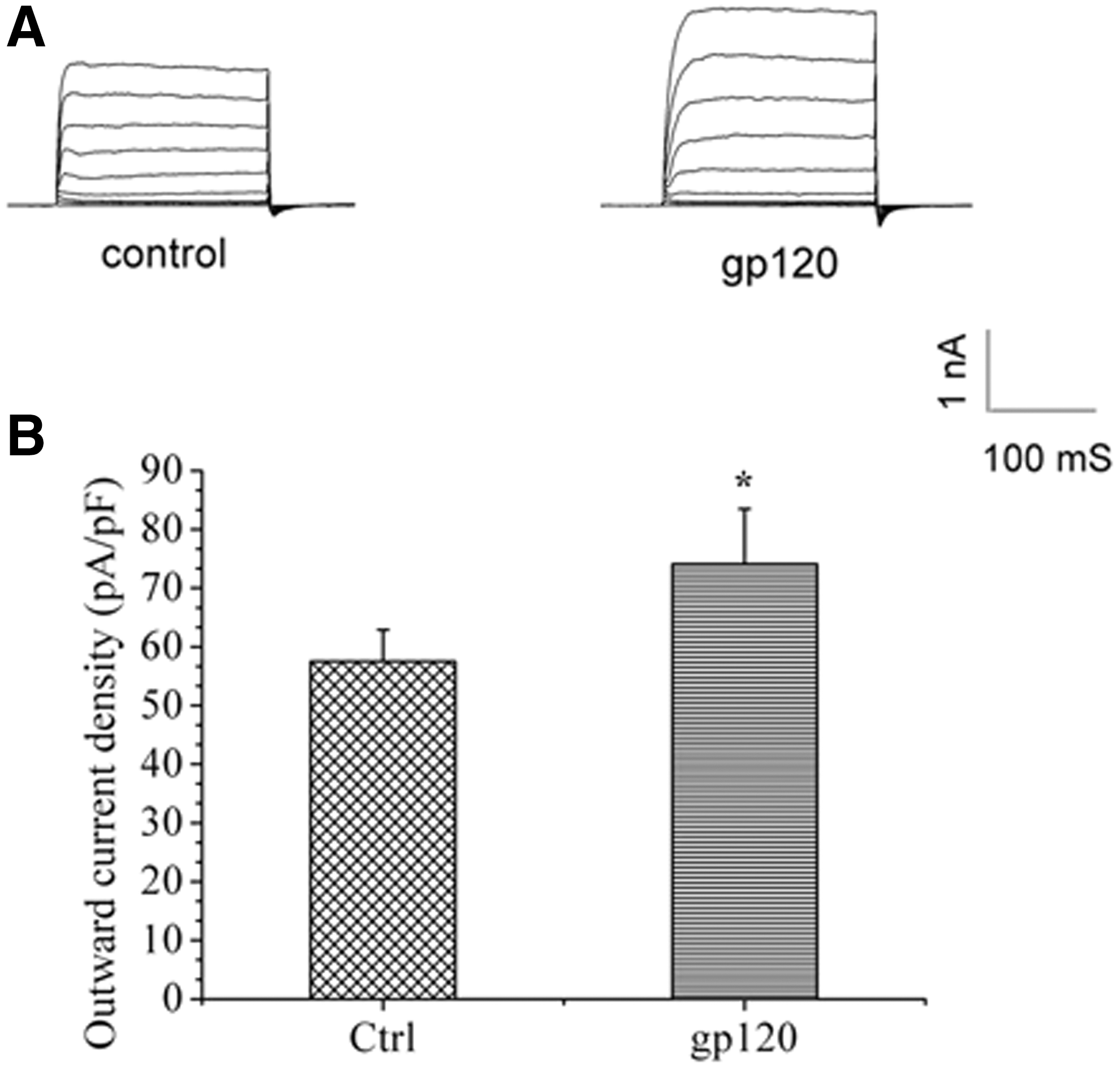
Envelope glycoprotein 120 (gp120) significantly enhanced potassium current (Ik). The typical example of Ik of hippocampal neurons were recorded in both the control and the gp120-treated groups (
Kv2.1 channel is one of the major components of the Ik in rat hippocampal neurons
After gp120 enhancement of the Ik was established, this study examined which subtype of the K+ channel might be involved. A previous study suggested that Kv2.1 is one major component (27) mediating Ik and is highly expressed in the hippocampus. Therefore, the Kv2.1-mediated Ik were investigated. The currents were evoked and recorded in the presence of 4-AP by a series of voltage-step pulses from −80 to 80 mV, while the holding potential was set at −80 mV. As shown in Figure 2, GxTX-1E, the blocker of Kv2.1, significantly decreased the outward K+ currents. For the 25 nM GxTX-1E-treated group, the K+ current density decreased to 67.7 ± 18.6% compared with the control group (100.0 ± 13.6%, n = 10; p < 0.01), whereas 50 nM and 100 nM GxTX-1E decreased current densities to 57.5 ± 9.54% and 60.6 ± 11.51%, respectively (n = 10, p < 0.01 compared to control; Fig. 2A and B).
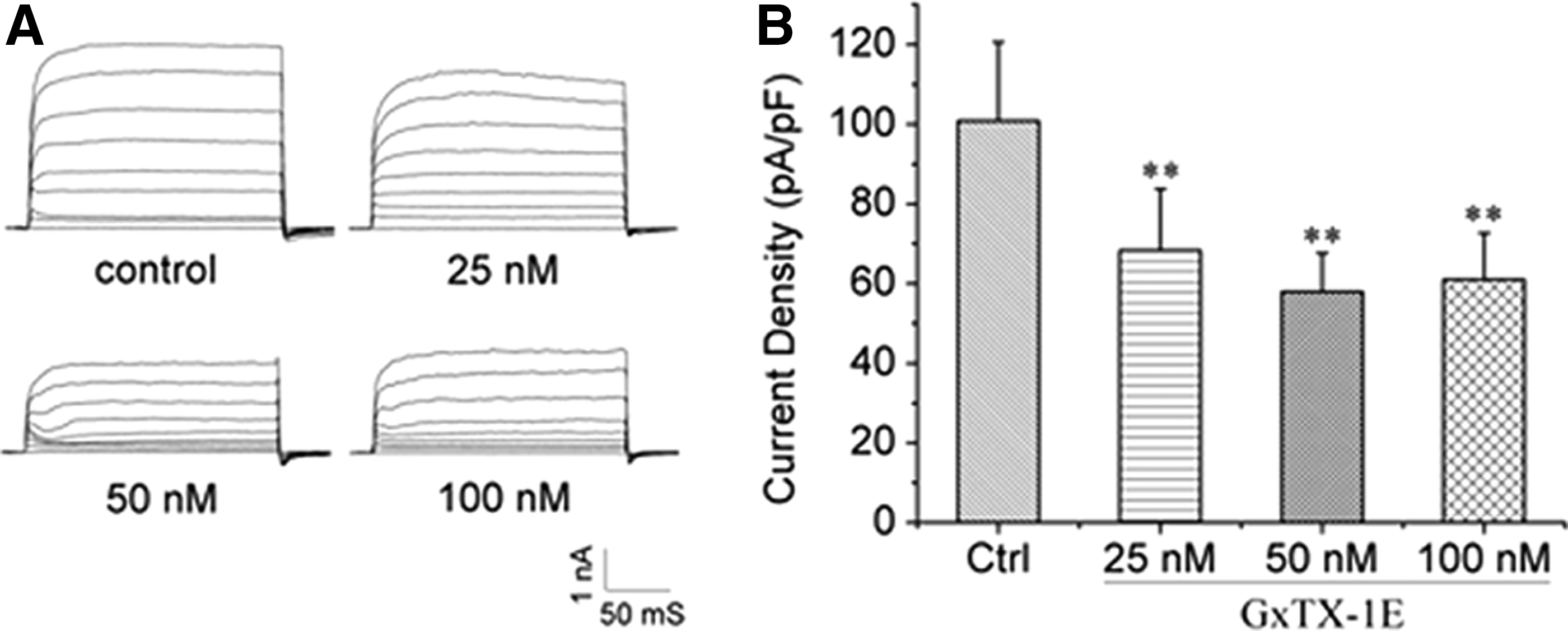
Kv2.1 was one of the major components of the Ik. The typical example of Ik was significantly inhibited by Kv2.1 blocker (GxTX-1E). The Ik was recorded in 25 nM, 50 nM, and 100 nM GxTX-1E containing solution (
gp120 upregulates Kv2.1 expression in hippocampal neurons
Having determined the importance of Kv2.1 in regulating the Ik, the next step was to examine whether the enhancement of the Ik induced by gp120 was associated with the upregulation of Kv2.1, since the channel expression was closely associated with the current density. Using Western blot assay, the results revealed that when the hippocampal neurons were exposed to gp120 (200, 500, and 1,000 pM) for 24 h, the protein level of Kv2.1 was significantly increased by 1.1-, 1.6-, and 1.5-fold in hippocampal neurons, respectively, compared with the control group (Fig. 3A and B). Next, the mRNA expression of Kv2.1 was determined by qPCR. It showed that gp120 significantly increased Kv2.1 expression in a dose-dependent manner (Fig. 3E). During the process, the protein expression of Kv2.2, another type of outward delayed-rectifier potassium channel, was also detected, which showed no significant change after being exposed to different levels of gp120 (Fig. 3C and D).
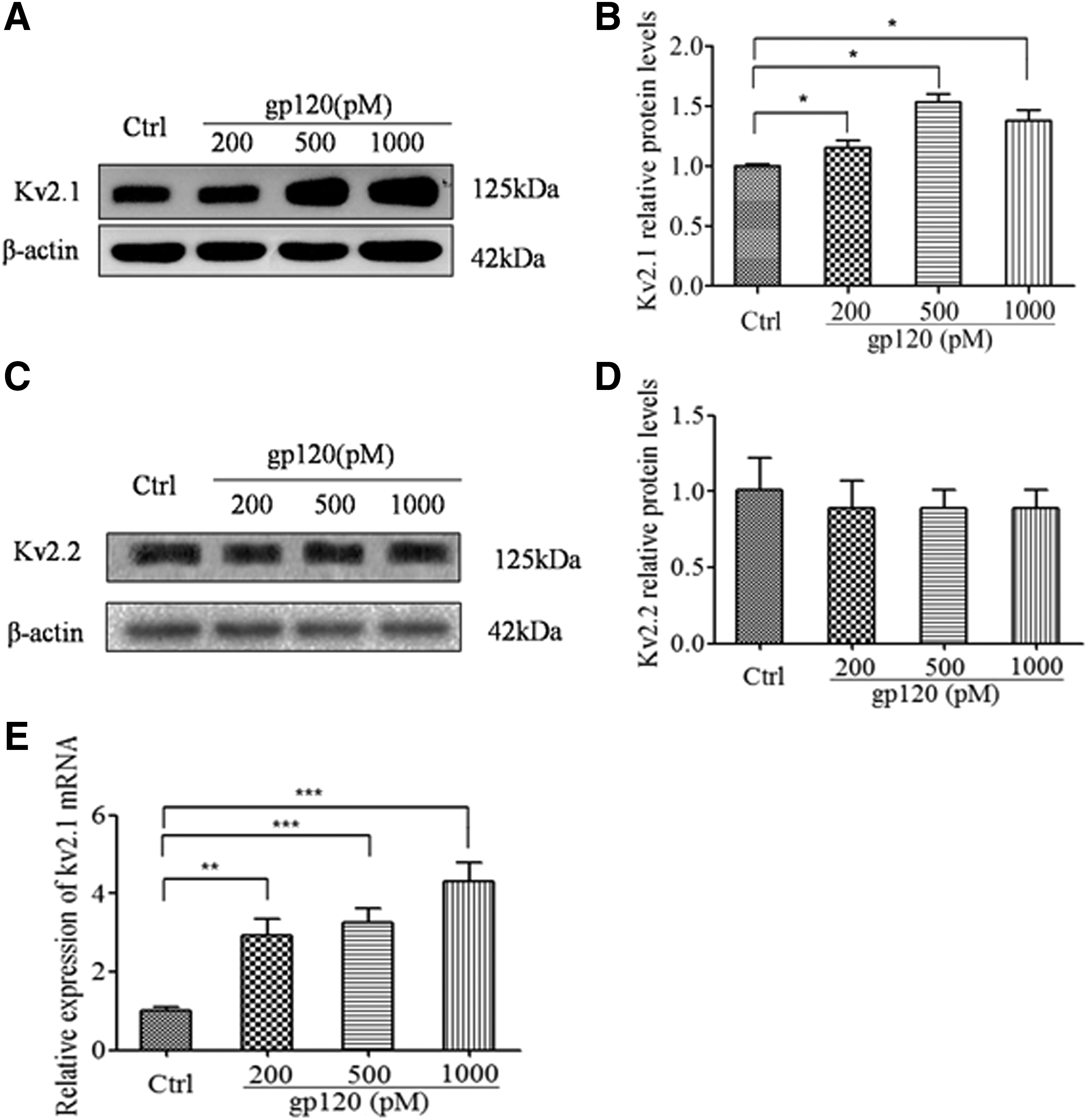
Effects of gp120 on Kv2.1 expression. Hippocampal neurons were treated with 200, 500, and 1,000 pM gp120 for 24 h. Then, levels of Kv2.1 and Kv2.2 protein were detected. β-Actin was used as a loading control (
Neuronal viability and apoptosis after gp120 treatment
To investigate gp120-induced neurotoxicity, the hippocampal neuronal viability was subjected to the MTT colorimetric assay after being incubated with varied concentrations of gp120 (100, 200, 500, 1,000, and 2,000 pM) for 24 h. As indicated in Figure 4A, gp120 exposure significantly decreased the neuronal viability in a dose-dependent manner (Fig. 4A), as 200, 500, 1,000, and 2,000 pM gp120 significantly reduced the neuronal viability by 14.32%, 19.79%, 28.37%, and 40.48%, respectively. To corroborate the findings that gp120-induced neural toxicity reproduced the reduction in cell viability, quantification of the active fragment of caspase-3 was subjected to Western blot. It revealed that gp120 facilitated caspase-3 activation, since an increase in the ratio of cleaved caspase-3 fragment to pro-caspase-3 was observed (Fig. 4B and C).
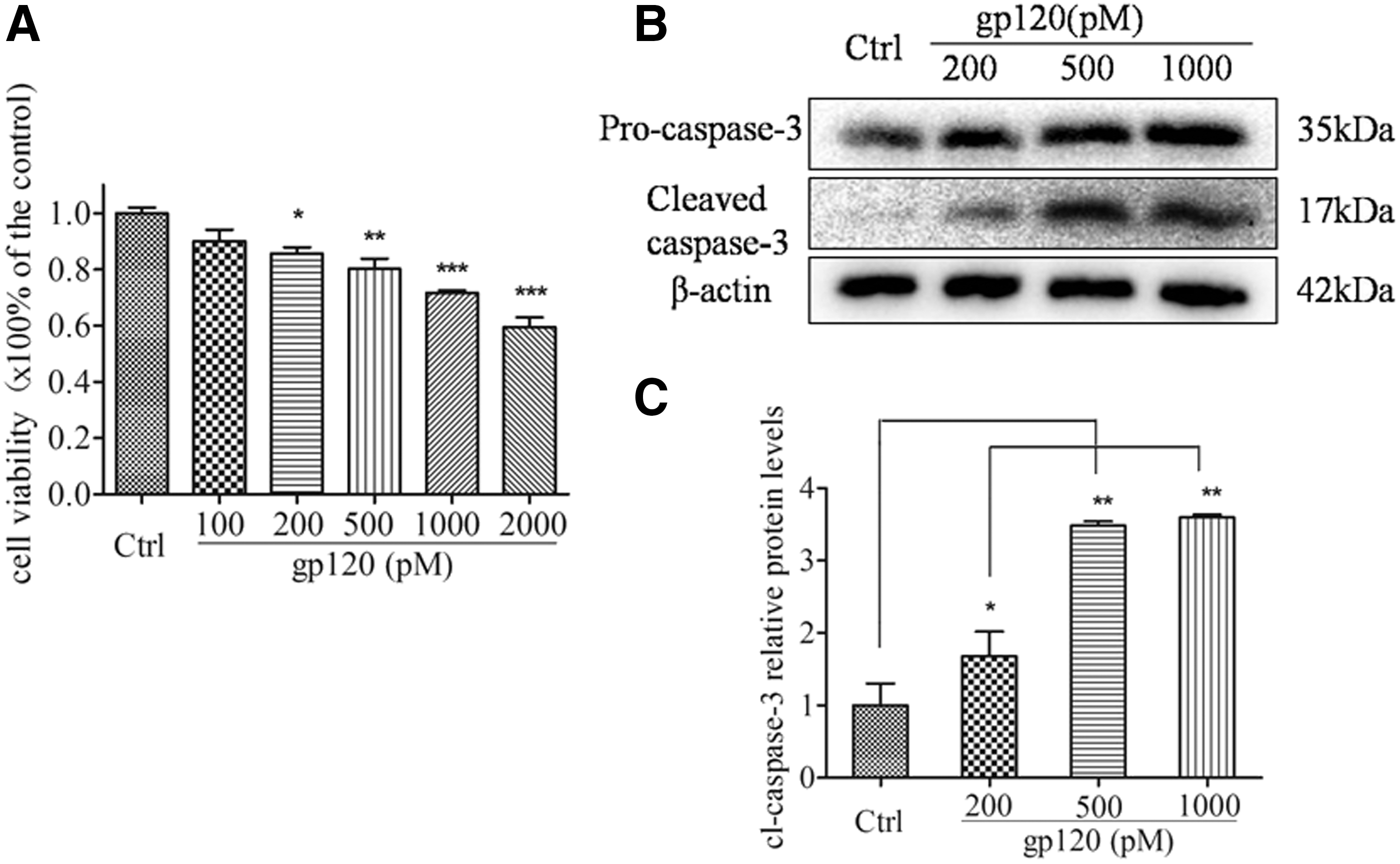
Gp120 decreased neuronal viability and triggered caspase-3 activation. The hippocampal neurons were incubated with 100, 200, 500, 1,000, and 2,000 pM gp120 for 24 h. The viability of neurons was analyzed by MTT assay (
Blockade of Kv2.1 reverses gp120-induced neuronal apoptosis and caspase-3 activation
The accumulating data suggested that the significant upregulation of Kv2.1 was closely associated with cell injury (31). Therefore, to determine whether Kv2.1 is involved in gp120-induced neural injury, the effects of GxTX-1E on gp120-induced neural apoptosis were examined by TUNEL staining. Cell nuclei were labeled with DAPI staining (blue), and apoptotic cells were determined by TUNEL staining (green; Fig. 5A). The results suggested that incubation of 1,000 pM gp120 led to remarkable neuronal apoptosis (40.5 ± 6.7%, n = 6) compared with the control group (4.7 ± 0.9%, n = 6). However, pretreatment of GxTX-1E (50 nM) significantly reduced gp120-induced neuronal apoptosis (6.9 ± 3.4%, n = 6; Fig. 5B). To confirm the effects of Kv2.1 on gp120 induced neural damage further, the caspase-3 expression was then detected by Western-blot. It was indicated that gp120 markedly facilitated caspase-3 activation, while pretreatment with GxTX-1E substantially attenuated the ratio of caspase-3 fragment to pro-caspase-3 (Fig. 5D), and caspase-3 activation was not affected when GxTX-1E was used alone (Fig. 5C).
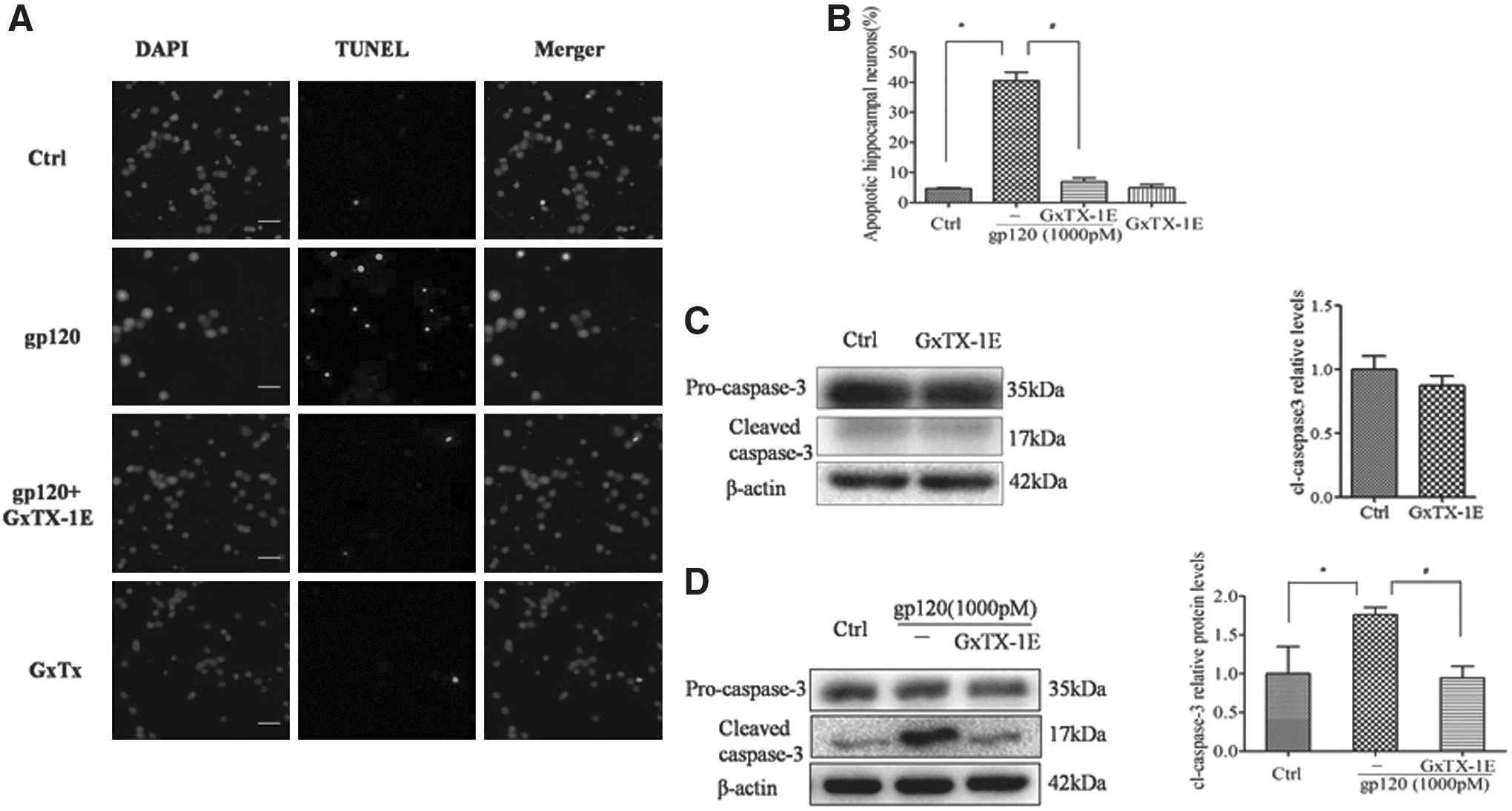
Blockade of Kv2.1 attenuated gp120-induced neuronal damage. Immunocychemical analysis of apoptosis (terminal deoxynucleotidyl transferase dUTP nick end labeling [TUNEL] staining) in hippocampal neuronal cultures induced by gp120 (1,000 pM) in the absence and presence of GxTX-1E (50 nM). Intact cell nuclei were visualized with DAPI staining of nucleic acids and apoptotic cells were labeled with TUNEL staining of fragmented DNA (
P38 MAPK rather than ERK1/2 and JNK is involved in gp120-mediated Kv2.1 upregulation
MAPK signal pathways were previously reported to be associated with HAND (24). Therefore, it was proposed that gp120 might exert its toxic effects through MAPK pathways. As shown in Figure 6, gp120 significantly activated the p38 MAPK pathway, and the level of phosphorylated p38 MAPK was obviously increased in the presence of 1,000 pM gp120 from 0.5 to 6 h (Fig. 6A). Moreover, the increment of Kv2.1 expression (Fig. 6C), as well as cleaved caspase-3 (Fig. 6E) mediated by gp120, was obviously downregulated by SB203580, the p38 MARK inhibitor, which did not affect Kv2.1 expression (Fig. 6B) and caspase-3 (Fig. 6D). Moreover, gp120 also activated the ERK1/2 (Fig. 7A) and JNK (Fig. 7B) pathways. However, the ERK1/2 inhibitor PD98059 and JNK inhibitor SP600125 failed to block the increment of Kv2.1 protein mediated by gp120 (Fig. 7D and F), and PD98059 and SP600125 did not affect Kv2.1 expression when they were used alone (Fig. 7C and E).

Involvement of p38 MAPK in gp120-mediated Kv2.1 and caspase-3 expression. Treatment with 1,000 pM gp120 increased phosphorylation of p38 MAPK in cultured rat hippocampal neurons from 0.5 h to 6 h (
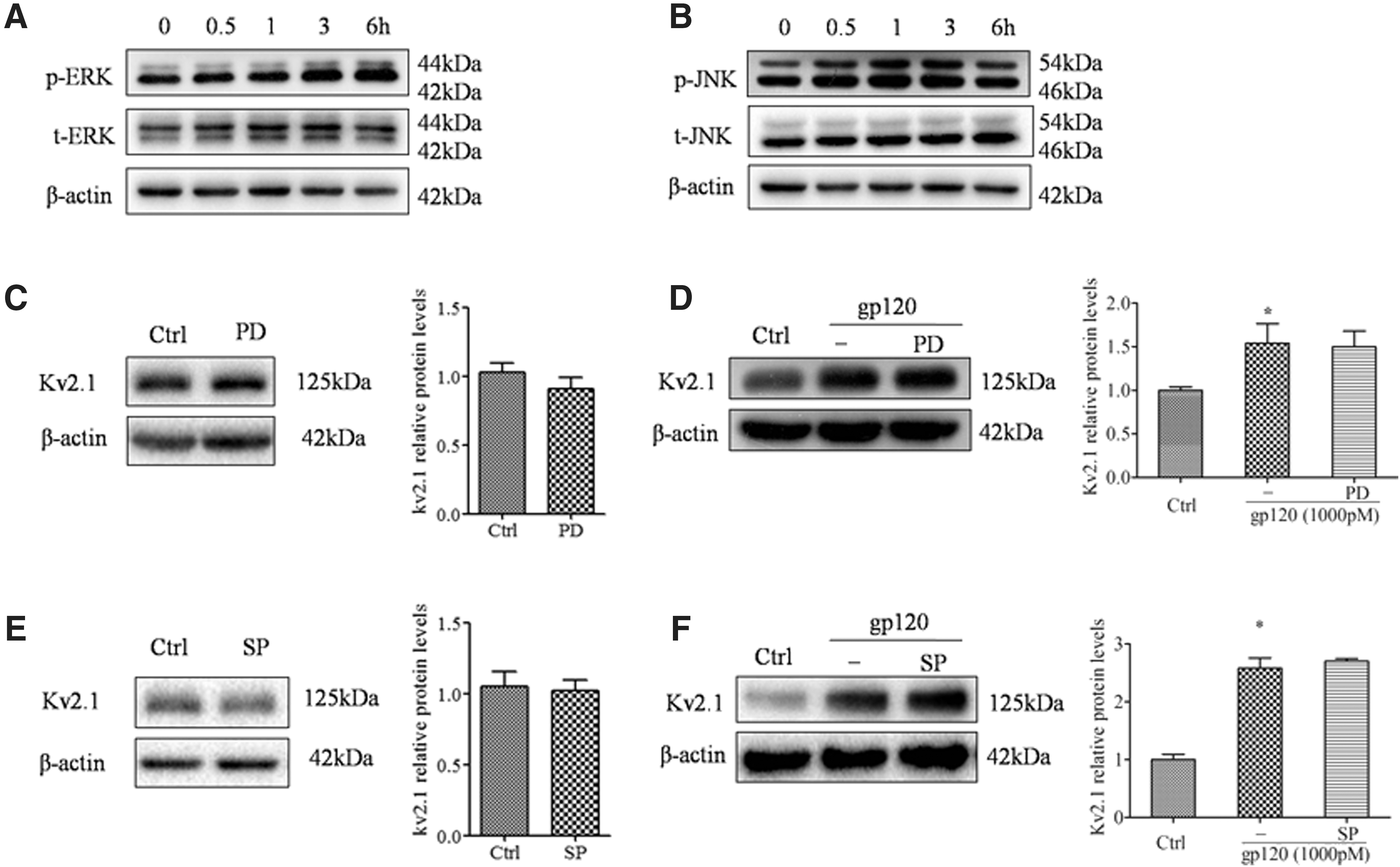
Gp120-activated ERK and JNK pathways. The p44/42 MAPK (ERK1/2) (
Discussion
Persistent HIV-associated neurodegenerative processes have been the concern of several neuropathological and neuropsychological studies (16). As one of the HIV envelope glycoproteins, systemic administration of gp120 in neonatal or adult rats resulted in deleterious effects on the neuron (37). Guyon and Yang demonstrated that gp120 was able to activate CXCR4 and NMDA receptors, resulted in enhancement of Ca2+ influx in neurons and finalized neurotoxicity (12,40). A similar result was also observed in cultures of rodent hippocampal neurons as well as in rodent brains in vivo (3). Thus, gp120 has been proposed to be the major etiologic agent inducing neuronal cell dysfunction or death and other debilitating neurological and behavior consequences of HIV-1 infection observed in HAND (15). Recently, the Kv channels attracted more attention because of their close association with cell injury (34). Several studies indicated that voltage-gated Ik was involved in gp120-induced neuronal apoptosis (5,18,21). Nonetheless, the neurotoxicity of gp120 on the hippocampus and which pathways are involved in gp120-induced hippocampal neuronal injury still need to be clarified.
The present study unambiguously shows the connections between gp120, MAPKs, and Kv channels, and two findings are presented. First, gp120-induced hippocampal neuronal injury might occur through the Kv2.1 channel, which manifests as outward K+ efflux and Kv2.1 upregulation. Second, the p38 MAPK pathway is involved in Kv2.1 upregulation and hippocampal neuronal injury. More recently, Shepherd et al. demonstrated that acute exposure (30 min) with a high dose of gp120 (10 nM) markedly activated Kv2.1, thereby enhancing the neuronal Ik current density via the Kv2.1 channel (35). In agreement with the aforementioned findings, the present study demonstrated that gp120 significantly enhanced the delayed outward rectified K+ currents in hippocampal neurons. However, the dose of gp120 used in the present study was even lower (ranging from 200 pM to 1 nM), and the incubation time was much longer (24 h) compared with Shepherd's work. Intriguingly, the two different treatments (30 min and 24 h) might contribute to the differential outcome. The acute exposure of gp120 (30 min) resulted in Kv2.1 activation, while 24 h exposure contributed to Kv2.1 upregulation but was not limited to current enhancement, implying that gp120-enhanced Ik current density might occur in two different ways. In addition, it is worth mentioning that gp120-induced K+ efflux, at least in the present study, even lasting for 24 h, which might contribute to neuronal injury and caspase-dependent apoptosis. Furthermore, gp120-induced neuronal injury is obviously attenuated by Kv2.1 blocker GxTX-1E, suggesting a specific role for Kv2.1 in neuronal injury induced by gp120. Kv2.2 is another type of outward delayed-rectifier Ik, which can also be blocked by GxTX-1E. In the present work, no significant changes were observed after gp120 exposure, which ruled out the possibility of Kv2.2 regulating caspase-dependent apoptosis.
P38 MAPK is currently reported to be closely associated with HIV replication in a variety of systems (11,13,38). HIV-induced hyperactivation of p38 MAPK leads to induction of biomarkers indicative of cellular exhaustion and apoptosis (8). Muthumani et al. found that HIV Nef mediated the apoptotic marker FasL (28) and the inhibitory receptor PD-1 (29) expression through activation of p38 MAPK pathway. Similar results by Leghmari et al. indicated that p38 MAPK was also involved in HIV Tat induced IL-10 production in monocytes (19). In line with the aforementioned discoveries, the present study demonstrated that HIV gp120 obviously activated the p38 MAPK pathway, upregulating the downstream target protein Kv2.1, and finalized cell damage. The data revealed that gp120 obviously induced upregulation of Kv2.1 and cleaved caspase-3, and these processes can be significantly attenuated by the blockage of p38 MAPK, which supported the association between Kv2.1 and cell death mediated by gp120 reported by Shepherd (34).
In summary, the present results provide and confirm the evidence that HIV-1 gp120 enhances the Ik might partially through Kv2.1 upregulation, and finalizes hippocampal neuronal apoptosis. In addition, the p38 MAPK pathway is indicated to be involved in gp120-induced Kv2.1 upregulation and neural injury. These results strongly suggest the enhancement of Ik mediated by gp120, which might occur through upregulation of Kv2.1, leading to hippocampal neuronal apoptosis via the p38 MAPK pathway. These findings, taken together, provide a potentially unique target for therapeutic intervention in HIV-1-associated or other neurodegenerative disorders that have symptoms of learning and memory ability decay. However, the possibility still cannot be ruled out that other K+ channel types are additionally involved in gp120-induced hippocampal injury. Furthermore, more work is required on the mechanisms for the enhancement of the K+ currents induced by gp120.
Footnotes
Acknowledgments
This work was supported by the National Science Foundation of China (81202230), the Priority Academic Program Development (PAPD), of Jiangsu Higher Education Institutions and the Foundation of Nanjing Medical University (2011NJMU275).
Author Disclosure Statement
No competing financial interests exist.