
Abstract
One of the key hallmarks of chronic human immunodeficiency virus type 1 (HIV-1) infection is the persistent immune activation triggered since early stages of the infection, followed by the development of an exhaustion phenomena, which leads to the inability of immune cells to respond appropriately to the virus and other pathogens, constituting the acquired immunodeficiency syndrome (AIDS); this exhausting state is characterized by a loss of effector functions of immune cells such as proliferation, production of cytokine, as well as cytotoxic potential and it has been attributable to an increased response of regulatory T cells and recently also to the expression in different cell populations of inhibitory molecules, such as programmed death receptor-1 (PD-1), cytotoxic T lymphocyte antigen-4 (CTLA-4), T cell immunoglobulin-3 (Tim-3), and lymphocyte activation gene-3 (LAG-3). The importance of these molecules relies on the possibility to restore the immune response once these molecules are blocked, constituting a potential therapeutic target for treatment during HIV infection. In this regard, we explored the available data evaluating the functional role of Treg cells and inhibitory molecules during the infection in both blood and gut-associated lymphoid tissue (GALT) and their contribution to the development of immune exhaustion and progression to AIDS, as well as their therapeutic potential.
Introduction
T
During the acute phase of infection, there is high viral replication with massive destruction of most of the effector memory T lymphocytes (CD4+CCR5+), located preferentially in the gut-associated lymphoid tissue (GALT) (10). Several immune mechanisms are then triggered, decreasing viral replication and allowing at least partial recovery of the number of the CD4+ T cells in peripheral blood but not in GALT.
The persistent depletion of CD4+CCR5+ T cells in GALT during the acute phase along with the apoptosis of enterocytes induced by viral proteins and the disjunction of intercellular bindings (11) lead to damage of the integrity of the mucosa membrane allowing translocation of bacterial products to the systemic circulation; this phenomenon triggers the immunological hyperactivation state, which is the main pathogenic mechanism during HIV-1 infection. The exacerbated activation is maintained during the chronic phase increasing the number of viral targets, leading to a progressive damage in secondary lymphoid organs, and finally, to the development of acquired immunodeficiency syndrome (AIDS) (65).
Despite being activated, immune cells do not fulfill their effector functions efficiently, allowing viral replication. One possibility is that these cells exhibit functional alterations or that mechanisms developed by the virus to evade the immune system exceed the effector response. In addition, an increased response of regulatory T cells (Treg) could also be favoring viral replication (62) as well as the expression of inhibitory molecules on different cell populations downregulating the immune response. In fact, upregulation of several of these molecules during HIV-1 infection, in association with AIDS progression, has been described (35).
The aim of this review is to gather the latest information on the functional role of Treg cells and inhibitory molecules during HIV-1 infection and their contribution to the development of immune exhaustion and progression to AIDS.
Immune Exhaustion
Chronic activation of lymphocytes promotes cell survival, but with reduced ability to respond to antigenic stimulation. This phenomenon is called immune exhaustion (35) and plays a crucial role during chronic viral infections; this phenomenon has been described during chronic infections, such as HIV-1 (60) and hepatitis B (HBV) and C virus (HCV) (37), and during oncogenic processes (16).
During HIV-1 infection, the progression to AIDS has been associated with an immune hyperactivation state that gradually leads to the immune exhaustion phenomenon, with loss of effector functions of T cells such as proliferation, production of cytokine, as well as cytotoxic potential, which together promote viral replication and persistence (35). The mechanisms underlying the development of this anergic state are currently being studied.
Treg Cell Functions and Their Association with the Immune Exhaustion State
Given their suppressive function, regulatory T cells could play an indirect but important role in the development of the immune exhaustion phenomenon by inhibiting specific immune responses (20,51). These cells are characterized by the constitutive expression of the alpha chain of the IL-2 receptor (CD25) and the forkhead box P3 transcription factor (FoxP3) that induces all the genes required for immune regulation (62). Treg constitutes a unique lineage, whose main function is suppressing the immune effector activity, promoting peripheral immune tolerance, and protecting the tissues from damage induced by excessive cell activation and proliferation (62). These cells need to be activated through the T cell receptor (TCR) to become suppressive implying that Treg activation is antigen specific (15); however, after activation, their function is accomplished by different mechanisms such as soluble factors like the regulatory cytokines TGF-β and IL-10, by cell–cell contact-dependent mechanisms or through the selective uptake of IL-2, which suppress the activation of both innate and adaptive immune cells that may or may not have the same antigen specificity that the Tregs have (62).
During oncogenic processes and chronic viral infections, the effects of a robust response of Treg may be detrimental, decreasing the specific immune responses, contributing to tumor and virus persistence. In breast, ovarian, and prostate cancer, an increased frequency of Treg infiltrating the tumor tissue, with high ability to suppress the proliferation and response of CD4+ T cells, has been reported; in fact, their presence was associated with the worst prognosis (6,17,45). During HBV infection, the peripheral expansion of this cell population was correlated with high viral load and decreased response of HBV-specific CD8+ T cells (63).
In the context of HIV infection, persistent immune activation, as well as viral particles, through gp120, induces Treg expansion (14); however, the role of these cells during the infection remains controversial depending on the stage of infection. During the acute phase, it has been postulated that an increase in the Treg number is related with suppression of excessive immune responses and lower damage to subjacent tissues (29); however, in the chronic phase, an increased frequency of these cells may favor viral replication by inhibiting HIV-1-specific responses as it was previously shown by others, including us (56). The impairment of HIV-specific CD4+ T cell responses by IL-10 was reported (40); in addition, Treg cells can also suppress the citolytic and nonlytic activity of HIV-specific CD8 T cells (36). An increased frequency of these cells in HIV-1-infected individuals has been also associated with failure in the immune reconstitution during the antiretroviral therapy (27).
Although there is no evidence of a direct effect of Treg inducing immune exhaustion, one possibility is that through the production of IL-10, they can influence the function of antigen-presenting cells (APC), such as dendritic cells (DCs), decreasing the expression of major histocompatibility Complex-II (MHC-II), costimulatory molecules, and inflammatory cytokines (19). These tolerogenic DCs induce a state of anergy in T cells with increased expression of cytotoxic T lymphocyte antigen-4 (CTLA-4) (64).
Inhibitory Receptors and Their Role in the Development of Immune Exhaustion
Immune exhaustion could be partially responsible for functional alteration of immune cells during chronic infections, including HIV-1 infection. Different studies indicate that the expression of these molecules is reversible, underlying the importance of studying them as possible therapeutic targets for the control of tumor processes and chronic viral infections. We will review here the most recently described exhausting molecules.
Programmed death receptor-1
This is a monomeric surface glycoprotein with an extracellular domain of the immunoglobulin superfamily (IgSF) linked to a transmembrane domain and a cytoplasmic domain with two signaling motifs based on tyrosine. Programmed death receptor-1 (PD-1) belongs to the family of CD28 molecules that together with molecules of the B7 family is involved in regulating the balance between inhibitory and costimulatory signals, influencing the immune response to infectious agents and the maintenance of immune tolerance (36).
This molecule is expressed by CD4+ and CD8+ T cells, NK cells, NK cells with invariant TCR (iNKT), B lymphocytes, and monocytes. Once cell activation occurs, PD-1 binds to one of the following ligands: (i) PD-1L (B7-H1) widely and constitutively expressed by hematopoietic and nonhematopoietic cells, such as B and T lymphocytes, DCs and mesenchymal stem cells, and/or (ii) PD-2L (B7-DC), which is not constitutively expressed but can be induced exclusively on DCs and inflammatory macrophages (36) by activation of the signal transducer and activator of transcription 6 (STAT-6) signaling pathway using stimulation with IL-4 (44).
The inhibition of T cell responses is given by PD-1/PD-1L grouping that occurs after the recognition and activation of the TCR. Both tyrosine signaling motifs in the cytoplasmic domain of PD-1 are phosphorylated leading to recruitment of SHP-2 and then of SHP-1 phosphatases; these phosphatases initiate dephosphorylation of molecules proximal to the TCR, blocking the signaling cascade mediated by TCR and CD28, thus suppressing activation of T cells (75).
In DCs, the interaction of PD-1 with its ligands decreases the expression of the maturation markers, CD40, CD80, and CD86, and increases IL-10 production, promoting a tolerogenic phenotype in DCs (39).
Furthermore, the PD-1/PD-1L interaction also contributes to the maintenance of a regulator profile through de novo generation of Treg from naive CD4+ T cells and the induction of a sustained expression of FoxP3 in this cellular population, increasing its suppressor function (22).
Cytotoxic T lymphocyte antigen-4
This transmembrane protein of the CD28 family is expressed as a homodimer that competes with the costimulatory molecule CD28 for binding to CD80 and CD86 molecules (B7 family) and does so with high affinity (12).
CTLA-4 is constitutively expressed in Treg and is induced by activation on other T cells. This molecule is localized in intracellular compartments and during the activation of the TCR and the cross-link of costimulatory molecules; it is carried to the cellular surface by the ADP ribosylation factor-1 (ARF-1) and the phospholipase D (PLD), promoting its exocytosis and expression at the cell membrane (58). Once interaction between CTLA-4 and CD80 or CD86 has occurred, signaling through TCR is inhibited avoiding the activation and proliferation of T cells and the production of IFN-γ and IL-2 cytokines (12). On DCs, this interaction leads to an increase in the expression of indoleamine 2,3-dioxygenase (IDO) involved in tryptophan catabolism reducing cellular proliferation and inhibiting the cytotoxic ability of T cells (7,22). The increase in IDO expression generates a favorable environment for Treg polarization, maintaining the suppressor environment (56).
T cell immunoglobulin-3
This molecule belongs to the family of Tim molecules; in mice, there are eight recognized members, but only three are expressed in humans. This molecule is mainly expressed on CD4+ and CD8+ T cells differentiated to Th1 or to a cytotoxic profile, respectively (46). Galectin-9 (Gal-9), also expressed on T cells, has been proposed as a possible ligand of T cell immunoglobulin-3 (Tim-3) and it has been related to the induction of regulatory cells (2,38,61); its expression is increased in the presence of IFN-γ, inducing the death of cells producing this cytokine through the caspase-1 pathway (31). Indeed, the interaction of Gal-9 with Tim-3, expressed on CD4+ and CD8+ T cells secreting IFN-γ, has been associated with the specific suppression of these T cells (78).
Although Tim-3 was considered an exclusive molecule expressed by Th1 T cells and cytotoxic T cells (CTL), its expression was later described in NK cells, in which it might negatively regulate the response of this cell population (48), and also in Treg, which has been associated with enhancement of their regulatory function, promoting tolerance (59). However, this molecule is not always involved in immune suppression; in DCs, the interaction between Tim-3 expressed on their surface with the ligand Gal-9 synergizes with the signaling induced by toll-like receptors (TLRs) promoting Th1 responses (1).
Lymphocyte activation gene-3
This is a surface receptor with inhibitory activity that belongs to the IgSF. It shares the same chromosomal location and a high structural homology with the CD4 receptor, thus binding to the major histocompatibility Complex-II (MHC-II) molecules with great affinity, acting as a negative competitor for this receptor (4).
Lymphocyte activation gene-3 (LAG-3) is expressed as a weak dimer on the surface of activated CD4+ and CD8+ T cells, and of γ/δ T cells and NK cells; recently, it was also reported on pDCs. Its expression is dependent on activation, and it is induced by cytokines such as IL-2, IL-7, and IL-12 (41).
The binding of LAG-3 to MHC-II results in inhibition of the TCR signaling by inhibition of the calcium influx. Decrease in the intracellular calcium leads to reduction in calcineurin phosphorylation; thus, the transcription of the IL-2 gene is not promoted, resulting in less proliferation of T cells and lower cytokine production (25).
In murine models, the expression of LAG-3 induces suppression of T cells and a functional exhaustion of CD8+ T cells in persistent infections (41). In natural and induced Treg, the expression of LAG-3 appears to be required to induce strong functional activity of this cell population (28). However, currently, there are few studies evaluating LAG-3 in humans.
In addition to the inhibitory role of LAG-3 described in T cell responses, on monocytes and DCs, the binding with MHC-II stimulates the production of TNF-α and IL-12 (3); it also prevents cell death mediated by FAS through the activation of the MAPK/Erk and PI3K/Akt pathways (26).
Despite the functional descriptions of these inhibitory molecules, the mechanisms and signaling pathways that regulate their expression still remain ambiguous. Recently, a pathway involved in the expression of these and other inhibitory molecules was proposed. Using an in vitro model of HIV infection in DCs, increased activation (phosphorylation) of STAT-3 mediated by P38 mitogen-activated protein kinase (MAPK) was associated with upregulation of these inhibiting receptors; likewise, blockade of this pathway decreased the expression of these inhibitory molecules restoring T cell proliferation (13). Specifically for PD-1, its induction in CD4 and CD8 T cells requires nuclear factor of activated T cells (NFAT) after TCR stimulation, while in macrophages, it occurs by signaling through the TLR/NF-κB pathway (5).
Expression of Inhibitory Molecules During HIV-1 Infection
During HIV-1 infection, both PD-1 and CTLA-4 have been the main inhibitory molecules described in the development of the state of immune exhaustion. In fact, high expression of PD-1 in CD4+ and CD8+ T cells has been correlated with markers of progression to AIDS (high viral load and low CD4+ T cell count) (18) and with reduced production of IFN-γ and perforin by specific CD8+ T cells in individuals with active infection, making these cells more susceptible to both spontaneously and FAS-mediated apoptosis (52,76). After starting antiretroviral therapy, the expression of PD-1 is substantially reduced, improving the quality of the immune response (18).
A low frequency of HIV-1-specific CD8+ T cells expressing PD-1 was reported in individuals controlling viral replication known as long-term nonprogressors (LTNP) (76), supporting the hypothesis that this molecule is associated with the progression to AIDS by suppression of the specific response against the virus and other pathogens.
Although the mechanisms through which HIV-1 infection induces the expression of PD-1 remain unclear, a recent report indicated that the viral protein Nef increases the expression of PD-1 on infected peripheral blood mononuclear cells from healthy donors, using mechanisms dependent on P38 MAPK activation, suggesting that this pathway could be involved (47).
In addition to T cells, PD-1 is also increased in other cell populations during HIV-1 infection, such as NK cells, limiting their ability to proliferate and affecting their effector function during the chronic phase of infection (50). In B cells, despite their activated phenotype, they present low proliferative ability and decreased ability to respond to antigens (49).
The ligand for PD-1, PD-1L, is also overexpressed in myeloid DCs inducing downregulation of the maturation markers, CD40, CD80, and CD86, altering the functionality of these cells and correlating with higher viral load and lower CD4+ T cell counts (73). In vitro exposure to HIV-1 of CD4+ CCR5+ T lymphocyte and monocytes from healthy donors increases the expression of PD-1L, which is IFN-α dependent at least in monocytes (8). This is an interesting finding, considering that high levels of IFN-α were found in plasma and lymphoid tissues of HIV-1-infected individuals, which could contribute to the immune exhausting state (8). The expression and effects of PD-1 and PD-1L on different cell populations are summarized in Figure 1.
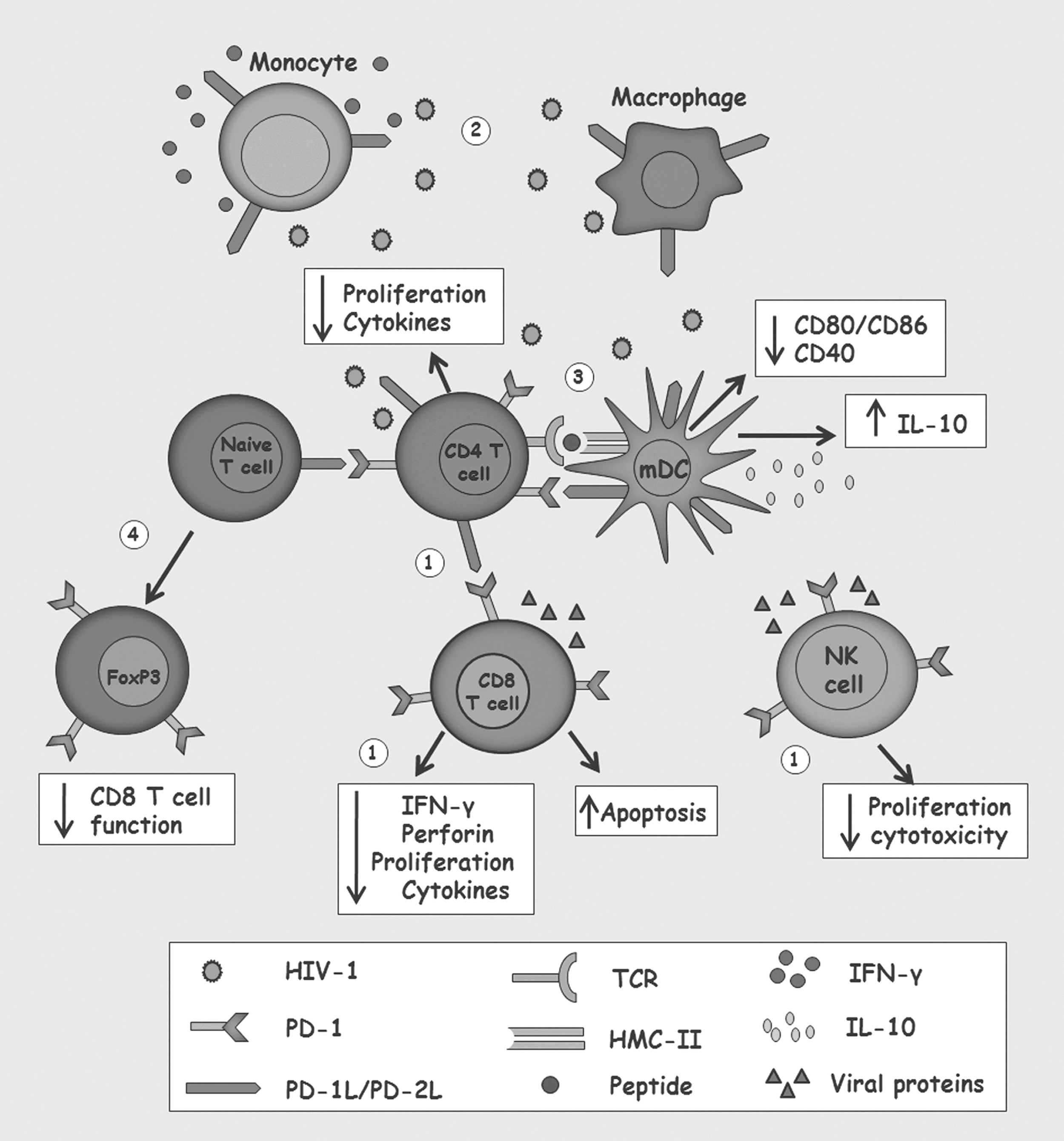
Alterations induced by the interaction of programmed death receptor-1 (PD-1) with PD-1L or PD-2L during human immunodeficiency virus type 1 (HIV-1) infection.
During HIV infection, increased expression of CTLA-4 on CD4+ T cells, which is negatively correlated with CD4+ T cell count and positively with progression to AIDS, has been reported. Indeed, CTLA-4 expression was increased in HIV-1-specific CD4+ T cells from HIV-1 progressors but not in LTNP individuals. The same study found that CD4+ T cells with high expression of CTLA-4 produced only IFN-γ, suggesting a monofunctional profile, while cells with lower expression showed a polyfunctional profile with production of both IL-2 and IFN-γ (32,33). In addition, it was observed that once the antiretroviral therapy is initiated, there is a decreased expression of this molecule with a simultaneous increase in the CD4+ T cell count and a decrease in virus to undetectable levels (42). However, taking into account that this molecule is constitutively expressed in Treg to perform their suppressive function (12) and that during the infection there is an increase in the frequency of these cells (56), it is plausible that during HIV infection the increase in CTLA-4 expression is also mediated by a higher percentage of Treg.
In addition, it has also been suggested that CTLA-4 may contribute to the pathogenesis of HIV-1 infection by increasing the expression of the CCR5 coreceptor, enhancing the cellular susceptibility to the infection by R5 strains (54). Furthermore, HIV infection of DCs, monocytes, and T cells induces the expression of costimulatory molecules of the B7 family, increasing the available ligands for CTLA-4 (33). A summarized model of CTLA-4 expression and effects is shown in Figure 2.
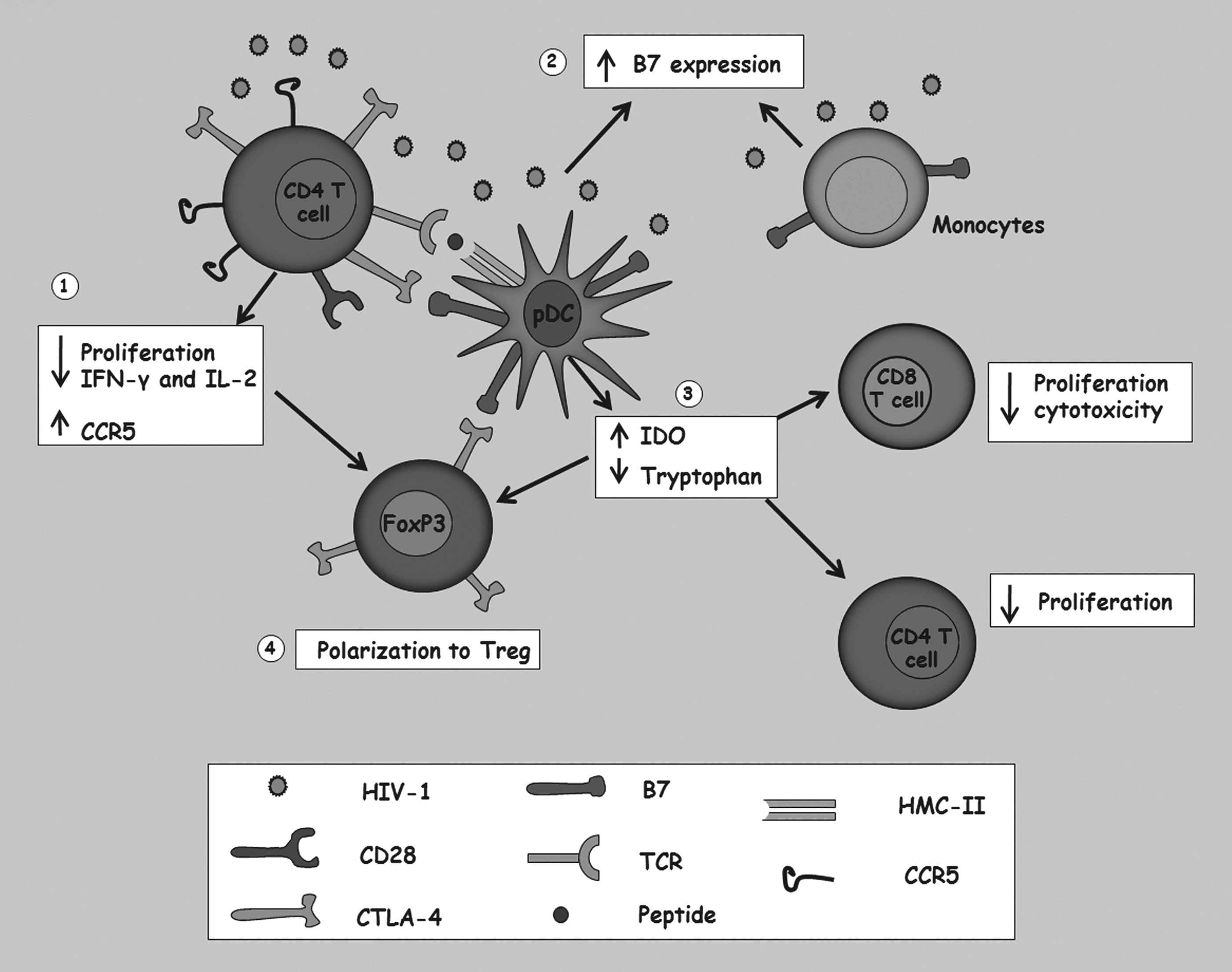
Alterations induced by interaction of cytotoxic T lymphocyte antigen-4 (CTLA-4) and its B7 receptor on the immune system, and its implication during the HIV-1 infection.
During HIV-1 infection, Tim-3 has been associated with lower proliferation and cytotoxicity and loss of polyfunctional T cell responses (30,57); in fact, the expression of this molecule is upregulated since early infection, and its coexpression with PD-1 correlates with a more exhausting state of T cells (77).
The expression of the described ligand for Tim-3, Gal-9, is also rapidly increased in plasma during the acute infection and remained elevated in the chronic phase, despite the viral suppression induced by antiretroviral therapy or by the spontaneous control observed in elite controller individuals; these results suggest that the interaction between Tim-3:Gal-9 remains active since early infection contributed to persistent T cell dysfunction (68) (Fig. 3).
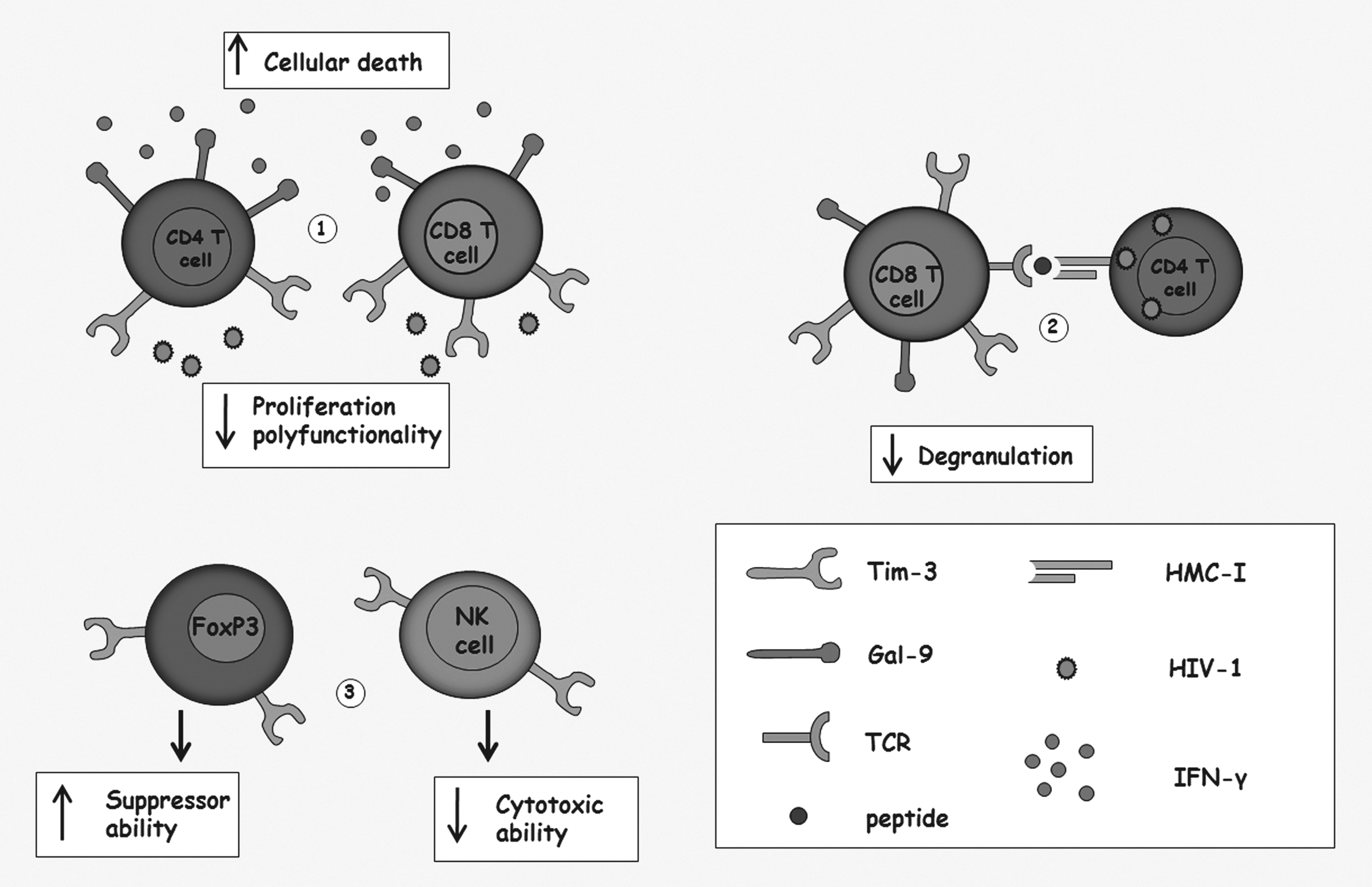
Alterations induced by the interaction of T cell immunoglobulin-3 (Tim-3) with its ligand Galectin-9 (Gal-9) on the immune system and its role during the HIV-1 infection.
LAG-3, as also the previous molecules, has attracted interest due to its contribution to the development of the immune exhaustion observed during HIV-1 infection; regarding this molecule, the data reported so far are contradictory. One study demonstrated a positive correlation between LAG-3 expression and viral load in patients with therapeutic failure (53). Moreover, microarrays studies of gene expression during this infection indicated a positive modulation of this molecule; specifically, the transcript levels of LAG-3 were significantly higher in fast progressor individuals compared to viremic nonprogressors (55).
In T cells, there is an upregulation of LAG-3 during the infection leading to reduction of T cell responses that correlated with disease progression (69). Taborda et al. also found a higher expression of LAG-3 as well as PD-1 and Tim-3 in NK cells from HIV progressor individuals compared to those who exhibit a spontaneous control of the infection (67).
In contrast, other authors did not find higher expression of LAG-3 during HIV-1 infection (43). Furthermore, HIV-1-specific CD8+ T cells that were exhausted (characterized by low cytokine production and proliferation) were also negative for this molecule (74).
The contribution of these molecules to the exhaustion state observed during HIV-1 infection affects mainly the adaptive immune response, specifically mediated by T cells; it raises the possibility that there are viral mechanisms that increase the expression of these molecules to avoid the immune control. However, further studies are required to confirm this hypothesis.
Markers of Immune Exhaustion in Gastrointestinal Mucosa and Lymphoid Tissues During HIV-1 Infection
As previously stated, the massive depletion of CD4+ T cells in the gastrointestinal lymphoid tissue during HIV-1 infection is the main mechanism responsible for the hyperactivation state and the development of immune exhaustion, due to changes in the integrity of the mucosa membrane and the translocation of microbial products into the systemic circulation system. For this reason, studies in GALT could give a clearer view of the alterations that occur during this infection and the contribution of the inhibitory molecules to the pathogenesis of the disease. However, studies concerning this issue are scarce. In chronically Simian immunodeficiency virus (SIV)-infected Rhesus Macaques, high expression of PD-1 in CD8+ T cells from GALT and other lymphoid tissues (80% and 50%, respectively) was reported (71); these authors also evaluated the potential of blocking this molecule in vivo to restore the immune response, finding a fast expansion of SIV-specific CD8+ T cells and an increase in the polyfunctionality of these cells with secretion of IFN-γ, IL-2, and TNF-α (72). In the sooty mangabey model, in which an early resolution of the infection with SIV is observed, there is an early induction of PD-1 in lymphoid tissue that gradually decreased; in fact, it has been proposed that such kinetics mediates the control of the immune hyperactivation response from the acute to the chronic phase of infection contributing to the asymptomatic state (21).
A study developed by our research group reported a higher expression of both PD-1 and CTLA-4 in CD4+ and CD8+ T cells from GALT of HIV-1-infected individuals, even after the instauration of the antiretroviral therapy. Furthermore, the increased expression of these molecules was correlated with higher viral load, suggesting a vicious circle between the virus and the persistent immune alterations during the chronic phase of the infection. This study also reported that the frequency of CD4+ T cells expressing CTLA-4 in gastrointestinal tissue was higher in highly active antiretroviral therapy (HAART) naive-infected individuals compared to treated individuals (56). No reports indicating the expression of Tim-3 and LAG-3 in GALT of infected individuals were found, pointing to the requirement to carry out such studies.
Therapeutic Potential of the Blockade of Inhibitory Molecules
All evidences recapitulated in this review demonstrate the suppressor ability of these molecules on the immune system and their implications in developing immune exhaustion in chronic viral infections; these findings highlight their importance as possible therapeutic targets, considering their reversible expression using blocking strategies.
There are ongoing phase I clinical trials, blocking the expression of PD-1 and PD-1L in hematologic malignancies, colon cancer, and lung cancer, which have led to promising results, even in patients at advanced stages of the disease (24,66).
In addition, blocking PD-1 during chronic viral infections has also led to interesting results; in individuals infected with HBV, in vitro blocking of PD-1 increased the survival of T cells and cytokine production, especially in cells from individuals coinfected with HIV-1 (63). In HIV-1 infection, in vitro blocking of both PD-1 and CTLA-4 in HIV-1-specific CD4+ and CD8+ T cells leads to recovery of the proliferation and to cytokine production (70).
As with PD-1, blocking of CTLA-4 with specific antibodies such as ipilimumab or tremelimumab (used in phase II clinical trials) induced sustained activation and proliferation of specific T cells against tumors (12). Similarly, during HIV-1 infection, in vitro blocking of this molecule induced increased proliferation of HIV-1-specific CD4+ T cells (32).
Assays blocking Tim-3 in cells from HIV-1-infected individuals also induced restoration in the proliferation ability and cytokine production of specific T cells (30); furthermore, a potent degranulation of cytotoxic T cells improving their ability to eliminate the virus and suppress CD4+ T cell infection was observed (57).
For LAG-3, the blockade of this inhibitory molecule significantly augments HIV-specific CD4+ and CD8+ T cell responses (69).
Conclusion
During HIV-1 infection, GALT is the main lymphoid tissue affected with a massive loss of CD4+ T cells that triggers a persistent and overwhelming state of immune activation; however, despite this hyperactivation, the immune system is unable to eradicate the virus and instead progresses to a state of immune exhaustion, favoring HIV-1 persistence. Consequently, inhibitory molecules have gained great interest based on their role during the development of this exhaustion state and particularly taking into consideration their reversible nature through the blocking of their activity, supporting their potential use as therapeutic targets. Furthermore, it has become evident that, while these molecules effectively suppress the response of several cell populations, their main target of action is the HIV-1-specific T cells, suggesting that positive regulation of these molecules could be induced as a mechanism to avoid the specific immune response.
Finally, considering that GALT is the main organ involved in the pathogenesis of HIV-1 infection, studies evaluating the expression of these molecules in this tissue are more than necessary to understand the viral mechanisms of immune evasion that lead to AIDS progression.
Footnotes
Acknowledgments
The authors thank Anne Lise Haenni for reviewing the article and for her constructive comments. The authors also thank the scholarship program “Jovenes Investigadores e innovadores–Colciencias 2013.” Financial support provided by CODI-Universidad de Antioquia, Convocatoria Programática 2012; Universidad de Antioquia UdeA.
Author Disclosure Statement
No competing financial interests exist.